
When a sterile medical device fails in surgery because its packaging couldn't maintain sterility during shipping, patient safety suffers first. Product recalls and damaged company reputation follow close behind.
Introduction to Packaging Validation
Medical device packaging validation proves your packaging systems protect device sterility from manufacturing through point of care. Your packaging serves as a sterile barrier system that maintains microbial protection throughout sterilization and distribution, helping devices remain sterile for years before reaching patients.
ISO 11607 provides the international framework for validating these systems. Part 1 sets requirements for materials and sterile barrier systems used to package terminally sterilized medical devices. Part 2 provides validation requirements for forming, sealing, and sterile barrier system processes. Together, they ensure packaging is both capable of maintaining sterility and proven to perform under real-world manufacturing conditions. FDA Quality System Regulation (21 CFR 820) enforces parallel requirements through design controls and process validation mandates, making validation a compliance necessity rather than an optional quality measure.
Validation is typically led by quality assurance, yet real success depends on cross-department coordination. Many delays come from confusion over roles or gaps in expectations. When engineering, regulatory, and manufacturing groups contribute their expertise in sync, validation becomes far more efficient and reliable.
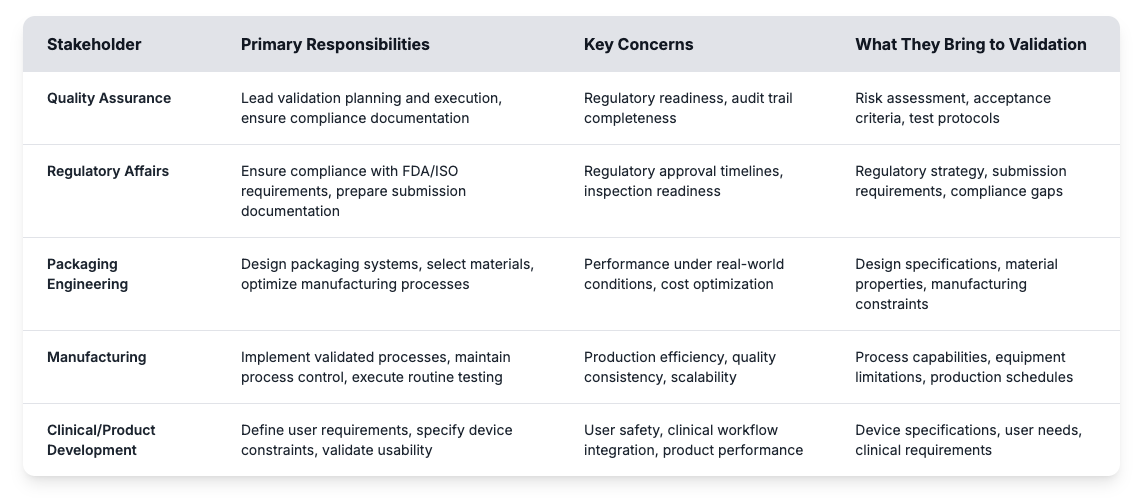
Getting these roles right helps medical device manufacturers build effective validation programs and avoid common pitfalls. Risk assessment identifies potential failure points while acceptance criteria establish measurable targets for packaging performance. Test protocols define how validation testing will prove that sterile barrier systems meet regulatory requirements throughout the intended shelf life. Building a successful validation program starts with understanding these foundational requirements and establishing clear processes that connect stakeholder input to measurable outcomes.
Foundational Requirements For Packaging Validation
Every successful validation program starts with three basic elements of packaging validation that regulators expect to see:
- Comprehensive validation plan: Your validation plan connects design decisions to compliance documentation. It defines who does what, when testing happens, and how you'll document results. Regulators look at these plans during inspections to see if your approach makes sense or if you're just checking boxes. Good plans include clear protocols with responsibility assignments plus realistic timelines that account for potential delays.
- Structured risk assessment: Risk assessment helps you focus validation resources where they matter most. You'll identify scenarios where packaging could compromise device sterility or safety - things like seal failures during shipping, material problems with your sterilization method, or packaging that's too difficult for users to open safely. This assessment tells you which tests are critical and which ones are nice-to-have.
- Scientifically justified acceptance criteria: Acceptance criteria define what "good enough" looks like for your packaging performance. They need to be specific and measurable while staying tied to patient safety rather than manufacturing convenience. Instead of vague criteria like "acceptable appearance," you'll set targets for seal strength, leak rates, and sterility maintenance that make sense for your specific device and use conditions.
Weak foundations create validation problems that multiply as programs progress. Teams often discover gaps too late, when fixing them means starting over with testing that should have been included from the beginning. The domino effect is predictable: scattered testing leads to incomplete evidence, which creates regulatory questions that delay approvals and increase costs.
The Four Pillars of Packaging Validation
ISO 11607 breaks packaging validation into four interconnected pillars that work together to prove your packaging won't fail when it matters most. Rather than treating these as separate checkboxes, successful teams understand how each pillar feeds into the next to build comprehensive evidence that packaging protects device sterility throughout the device lifecycle.

You can't prove consistent packaging performance (PQ) without knowing your equipment operates reliably (OQ). You can't trust equipment performance without proper installation (IQ). Material qualification (MQ) runs alongside equipment validation to ensure everything works together as a complete packaging system. When done properly, this systematic approach builds validation evidence step by step with each pillar reinforcing the others. When you skip steps, you create expensive problems:
- Skipping IQ means you don't know if equipment problems are messing up your later test results.
- Weak OQ leaves you with process parameters that look good in testing but fall apart under production stress.
- Shortcutting material qualification is how teams end up with packaging that fails during shipping or loses sterility over time.
Think of validation like building a house - you wouldn't start with the roof before laying a solid foundation. The four pillars work the same way, with each stage supporting what comes next. Skip the groundwork and you'll spend far more time and money fixing problems that could have been prevented with a systematic approach from the start.
Installation Qualification: Equipment Verification and Calibration
Without solid baseline documentation, equipment problems and process failures become impossible to separate during later validation stages. Installation qualification (IQ) prevents this by establishing exactly what you're starting with before any performance testing begins.
The core deliverable from IQ is baseline parameter documentation that captures your equipment's starting point. You'll record:
- Operating ranges for critical parameters like temperature, pressure, speed, and dwell time
- Default settings and control system configurations
- Environmental conditions in the packaging area
- Equipment performance characteristics under no-load conditions
- Preventive maintenance requirements and schedules that keep equipment performing consistently
Equipment verification and calibration establish the foundation for these baselines. You'll confirm that all components match purchase specifications, utilities meet requirements, and safety systems function properly. For sealing equipment, you'll document temperature and pressure capabilities along with heating element specifications. Control systems get verified to confirm they can maintain required parameters.
Calibration extends the foundation of IQ. Instruments that monitor or control the packaging process - temperature controllers, pressure gauges, timing systems, and testing tools - must have certificates traceable to national standards. These records anchor equipment performance and demonstrate process control during validation and routine production.
That record set also becomes the reference point for later stages. If seal strength drifts during performance qualification, results can be traced back to equipment variation documented in IQ. Without this level of documentation, distinguishing between equipment issues and process failures becomes difficult, leading to costly investigations that thorough upfront work would have prevented.
Operational Qualification: Testing Across Operating Ranges
Your equipment might run perfectly in ideal conditions but give inconsistent results when production gets messy. Operational qualification (OQ) reveals these trouble zones before they cause problems downstream. By testing parameters at their limits, teams can see where the process holds steady and where performance begins to break down.
Your goal is establishing process parameters that production teams will use daily - the operating limits with normal set points plus acceptable variation ranges. You'll find ranges where equipment produces reliable results consistently, with optimal settings that give the best performance and safety margins that prevent problems. Testing equipment functionality means systematically varying one parameter while holding others constant, then monitoring package quality outputs. Testing at the edges helps identify where performance starts breaking down. This becomes your safety margin for routine production.
OQ records need to go beyond listing what the equipment can do. They should also capture the conditions that keep results consistent. For example, high humidity may interfere with sealing, temperature changes can cause drift in critical parameters, and nearby machinery might introduce vibration that reduces precision.
This testing catches parameter combinations that would create problems in production before they actually show up. Your equipment might run at certain settings but give inconsistent results that cause headaches downstream. OQ identifies these trouble zones so production teams can steer clear, with the ranges you establish here becoming the foundation for operator training and process alarms that keep manufacturing running smoothly.
Performance Qualification: Proving Consistent Packaging Performance
Performance qualification (PQ) demonstrates that your complete packaging process consistently produces packages that meet all acceptance criteria under real production conditions. Here's where validation moves from proving capability to proving reliability.
The reality is that your packaging process needs to work when everything isn't perfect. Equipment settings drift slightly, operators change between shifts, and material lots vary in quality. Environmental conditions fluctuate throughout the day. PQ testing shows that your process still produces acceptable packages despite these normal variations. Worst-case scenario testing takes this further by pushing your validated process to realistic limits. Stress testing should reflect the realities of production, not just ideal conditions. That may mean running at maximum line speeds while operators push to meet deadlines, working under reduced environmental controls if HVAC systems aren’t performing well, or testing quality at the end of a shift when fatigue can influence results.
Running validation testing under actual production conditions means using the same materials, equipment settings, and environmental conditions that manufacturing teams face daily. Capturing natural process variation across time, operators, material lots, and equipment states matters more than testing under artificially controlled conditions that don't represent reality. Statistical planning sets the amount of testing needed to prove consistent performance. A well-designed sampling plan covers multiple runs and operators, ensuring results reflect real process capability instead of chance. That evidence then drives process monitoring and sustains validated performance during everyday manufacturing.
Materials and Design Qualification: Ensuring Compatibility and Protection
Material qualification (MQ) shows that device packaging materials can perform as intended across the full device lifecycle. For terminally sterilized medical devices, this means selecting primary packaging that withstands the chosen sterilization method and continues to protect sterility during distribution and storage. Packaging system validation then confirms that materials, design, and sealing function together as a sterile barrier throughout the device’s intended life.
When selecting materials, teams must evaluate three factors:
- The sterilization method that will be used
- The storage environment the package must withstand
- The user requirements for safe opening and device access
Each sterilization method presents unique challenges. Ethylene oxide sterilization typically requires materials that resist chemical degradation while allowing gas penetration and evacuation for effective sterilization and aeration. Gamma sterilization exposes packaging to radiation that may weaken strength and flexibility. Steam sterilization combines high temperature with humidity that can compromise barrier properties. If these risks aren’t addressed early, the sterile barrier can fail in real-world use, putting both compliance and patients safety at risk.
Material compatibility testing evaluates how your packaging interacts with device components over the entire intended shelf life, but it also needs to address practical usability. Materials must also maintain their microbial barrier properties to ensure sterile barriers continue blocking contaminants throughout the intended shelf life. At the same time, peel strength needs to be strong enough to maintain seal integrity during distribution but gentle enough for healthcare providers to open aseptically without tearing. You'll run accelerated aging studies that simulate years of storage in weeks, combined with real time aging under actual storage conditions to confirm your predictions hold up.
Material Testing Protocols:
Initial testing protocols help establish baseline material performance before full validation studies begin. Focus on designing test protocols that stress materials the way they'll actually be stressed in real-world use rather than meeting arbitrary test specifications that don't relate to actual performance requirements. Opening characteristics should allow for clean, controlled access that doesn't compromise sterility or scatter particles into the sterile field, which means your testing needs to simulate actual clinical use conditions rather than ideal laboratory environments.
Distribution and Transit Testing: Simulating Real-World Shipping
Packaging must withstand the full journey from the manufacturing floor to the patient’s bedside. Distribution testing recreates the stresses of shipping and handling so teams can confirm that sterile barriers stay intact and functional across the supply chain.
ASTM D4169 and ISTA protocols outline how to simulate distribution environments, but you need to tailor them to your actual shipping conditions. Instead of isolated checks, testing should reflect the full journey: a package may be dropped, rattled by transport vibration, or crushed under warehouse loads. Along the way, orientation shifts, repeated handling, and changing climate conditions all test the strength of the sterile barrier. Running these stresses in sequence gives a truer picture of how packaging performs in real-world distribution.
Packages go through several checks after distribution testing. Visual inspection looks for surface damage, while strength testing confirms that closures stay intact. More sensitive methods, such as bubble tests, uncover micro-leaks in sterile barriers that can’t be seen by the eye. The final step is integrity testing, proving the microbial barrier still performs even after simulated shipping - and those results only matter if the testing reflects real shipping conditions. Packages that pass generic protocols yet fail in the field lead to recalls while damaging company reputations. Most importantly, they put patients at risk.

Accelerated and Real-Time Aging Studies: Proving Shelf Life Claims
Shelf-life claims stand or fall on evidence that packaging preserves its sterile barrier for as long as devices remain on the market. Accelerated aging gives manufacturers an early view of packaging performance by using elevated temperature and humidity to simulate years of storage in just weeks. ASTM F1980 (current version) outlines the approach, but the real difficulty is designing conditions that mirror actual environments without introducing failure modes that would never happen in practice. When designed well, these studies produce preliminary data that supports initial shelf-life claims and helps products move forward while longer-term studies are still underway.
Real-time aging completes the picture. Packages from final production runs are stored under true conditions and monitored for performance over months or years. This evidence demonstrates how packaging withstands the day-to-day realities of storage and distribution, providing the confirmatory data regulators expect. Teams that begin real-time studies early avoid data gaps that can slow approvals or renewals later.
The most reliable shelf-life determinations come from combining accelerated projections with real-time confirmation. Statistical analysis shows when packaging properties drop below your targets and gives you confidence intervals for shelf life claims. Weak planning, however, can undermine the entire effort - testing prototypes instead of production runs, selecting storage conditions that don’t reflect reality, or underestimating sample sizes all erode credibility.
With the right approach, aging studies prove that packaging will protect patients across its intended lifespan while giving manufacturers and regulators the confidence that claims will hold over time. This evidence goes beyond regulatory requirements to demonstrate real-world performance that matters for patient safety.
Usability Evaluation and Human Factors: Validating User Interaction
A packaging system can meet every technical standard and still fail if it can’t be opened safely in the hands of a clinician. Usability validation ensures that sterile barrier systems hold up not only in the lab, but also in the real-world moment when devices are opened for patient care. Both FDA and ISO 11607 tie usability directly to risk management, recognizing that packaging that compromises aseptic technique is effectively a failed system.
A central focus is aseptic presentation testing - proof that the sterile barrier can be opened safely in practice. Closure systems and package seals must resist premature failure yet open smoothly under clinical conditions. Seal integrity is essential. Packages need to peel cleanly without tearing, and the design must keep particles or debris out of the sterile field. A frequent problem is the over-engineered seal - strong enough to meet test targets but so difficult to open that aseptic technique becomes impractical. Simulated use testing helps surface these issues before products reach the market.
In practice, testing often involves nurses or surgical technicians opening packages under sterile conditions while observers note any breaks in technique. Glove type matters: nitrile and latex provide different grip and feel. Other real-world factors - such as OR lighting, time pressure during procedures, or how well the package itself cooperates - also affect the ability to maintain sterility. Even details like peel tabs can make the difference: easy to manage with bare hands, but nearly impossible with gloves. Excessive seal strength poses the same risk, forcing workarounds that undermine the whole purpose of sterile packaging. A seal that can’t be opened aseptically doesn’t just frustrate users, it directly compromises sterility at the point of care.
Equally important is understanding how people interact with the package in practice. Usability studies observe healthcare providers opening devices under realistic conditions, surfacing moments of hesitation along with technique variation or potential contamination risk. These observations reveal problems that material testing misses - seals that demand excessive force or packaging that tears unpredictably when users need quick access. Unclear opening cues create confusion that compromises sterile technique during actual procedures.
Finally, validation must account for training requirements. If safe use depends on complex instructions, the risk of error rises. Assessing training needs early helps confirm that the packaging is intuitive for its intended users. Any instructions essential for safe handling should be documented clearly. Teams that take this step up front are better positioned to avoid costly redesigns or unexpected complaints after launch.
When usability is built into validation from the start, manufacturers gain more than compliance evidence. They deliver packaging that works for patients and clinicians alike, while assuring regulators it performs where it matters most.
Process Performance Qualification: Validating Manufacturing Consistency
Even the most carefully designed package fails if the manufacturing process can’t deliver the same result every time. Process Performance Qualification (PPQ) proves that packaging runs don’t just succeed in the lab or during validation trials, but remain consistent at full production scale. This is the point where both regulators and internal quality teams need proof that packaging systems can withstand real-world production.
A strong PPQ examines the entire production sequence, including assembly processes. It looks at how materials are stored, how operators handle components, and whether environmental controls keep conditions stable. Any weakness in those areas can erode packaging performance, even when equipment settings are dialed in.
Reliability comes from repeatability. That’s why PPQ requires multiple production runs to prove the process delivers consistent results across different batches. Teams typically validate three consecutive lots to show that performance is consistent across different production runs, though requirements may vary based on product risk and regulatory guidance. Ongoing monitoring then sustains that assurance, catching drift before it turns into quality issues.
The payoff extends well beyond compliance. A well-executed PPQ delivers benefits that reach far into daily operations. It makes production more predictable while accelerating release decisions. Most importantly, it builds confidence with regulators that the system is under control.
Revalidation Requirements and Change Management
Change is inevitable in manufacturing, and every change raises the question of whether your packaging system still does its job. Revalidation provides the evidence that it does, preventing small adjustments from creating large risks. Regulators view uncontrolled change as a red flag, and manufacturers should too. Common triggers include:
- Equipment updates, such as new sealing or inspection systems
- Sterilization changes, whether a shift in method or parameter adjustments
- Material modifications, from switching suppliers to selecting different substrates
- Process changes, even minor ones that alter operating conditions
The scope of revalidation depends on risk and documented validation requirements.Any change to a process raises the question of sterility. Teams may find that a small adjustment affects seal integrity. In other cases, material compatibility needs to be reassessed. Sometimes the concern is whether microbial barrier properties will still hold. Whatever the trigger, follow-up testing provides the proof that packaging continues to protect as intended. Regular reviews are also expected, since equipment wears over time. Shifts in suppliers or gradual process drift can have the same impact, and each must be monitored.
Good documentation pulls all of this together into evidence regulators can trust. It should explain the reasoning behind each test, not just capture that testing occurred. Clear records provide more than a paper trail. They give regulators confidence, help teams manage change proactively, and most importantly show that packaging continues to protect patients well beyond the first validation report.
Common Challenges and Effective Solutions
Validation can stumble for familiar reasons. Sample sizes are often underestimated, worst-case testing gets skipped, and documentation slips until it no longer reflects what was done. Teams also spend hours manually comparing documents and second-guessing whether they caught every critical change. Each of these missteps may feel small in the moment, but together they create costly rework and invite regulatory findings that damage credibility long after the audit ends. The manual review bottleneck is particularly problematic because it bogs down experienced quality professionals in tedious comparisons instead of applying their expertise to risk assessment and strategic decision making.
The stronger approach is to build validation as a strategic activity. Clear acceptance criteria set from the start help prevent late-stage disagreements, especially when quality, engineering, and regulatory teams align early in the process. When validation runs throughout development rather than being treated as a final step, gaps become easier to spot while regulatory reviews move more smoothly.
Working efficiently means putting effort where it counts most, not just cutting corners. Risk-based sampling focuses on resources where failure would matter most. Test plans can be designed to serve more than one requirement at a time, cutting redundancy without cutting rigor. Used this way, best practices save time while strengthening the evidence base, ensuring packaging earns regulator confidence and continues to protect patients in use.
Documentation and Regulatory Submission
For regulators, if it isn’t documented, it didn’t happen. Manufacturers face the same reality: incomplete records can slow approvals while triggering inspection findings that weaken trust in the quality system.
Strong documentation closes those gaps. A complete record shows how testing was planned and executed while connecting results back to acceptance criteria. This documentation demonstrates regulatory compliance. Certificates of validation and technical files then formalize this evidence, confirming that packaging systems meet both ISO 11607 and FDA expectations. More than a binder of reports, effective documentation weaves together protocols, data, and risk assessments into a controlled, inspection-ready story of compliance. When regulators ask, manufacturers need to show not just what was tested, but why, how, and with what outcome.
Thorough documentation doesn’t just satisfy regulators; it keeps devices moving to market without costly interruptions. When records are complete and inspection-ready, manufacturers can defend their process with confidence and patients receive packaging that protects them throughout the device lifecycle.
Ensuring Packaging System Success
Packaging validation proves that sterile barrier systems can withstand the journey from factory floor to point of care. Grounded in careful planning and backed by solid testing and documentation, validation gives manufacturers confidence their packaging will perform in the field and satisfy regulators. Treating validation as a lifecycle responsibility, not a one-time exercise, prevents costly failures and strengthens confidence across quality teams and regulators alike.
The landscape is evolving. Digital validation platforms and tighter links to risk management are reshaping best practices, while regulators move toward greater global alignment. What will not change is the goal: ensuring packaging maintains sterility so clinicians can rely on every device and patients receive the protection they deserve. Looking ahead, closer integration of packaging validation with sterilization validation will shape industry practice, ensuring sterility claims are supported end-to-end.
GlobalVision’s Verify accelerates packaging validation by automating document and artwork inspection, giving teams compliance confidence without slowing them down.
