

Lorsqu'un dispositif médical stérile échoue lors d'une intervention chirurgicale parce que son emballage n'a pas pu maintenir la stérilité pendant le transport, la sécurité des patients en pâtit en premier. Les rappels de produits et l'atteinte à la réputation de l'entreprise suivent de près.
Présentation de la validation des emballages
La validation des emballages des dispositifs médicaux prouve que vos systèmes d'emballage protègent la stérilité des dispositifs, de la fabrication au point de service. Votre emballage sert de système de barrière stérile qui assure la protection microbienne tout au long de la stérilisation et de la distribution, ce qui permet aux dispositifs de rester stériles pendant des années avant d'atteindre les patients.
NORME ISO 11607 fournit le cadre international pour la validation de ces systèmes. La partie 1 définit les exigences relatives aux matériaux et aux systèmes de barrières stériles utilisés pour emballer les dispositifs médicaux stérilisés au stade terminal. La partie 2 fournit les exigences de validation pour les procédés de formage, de scellage et de systèmes de barrière stériles. Ensemble, ils garantissent que les emballages sont à la fois capables de maintenir leur stérilité et ont prouvé leur efficacité dans des conditions de fabrication réelles. Réglementation du système qualité de la FDA (21 VOIR 820) applique des exigences parallèles par le biais de contrôles de conception et de mandats de validation des processus, faisant de la validation une nécessité de conformité plutôt qu'une mesure de qualité facultative.
La validation est généralement menée par l'assurance qualité, mais le véritable succès dépend de la coordination interservices. De nombreux retards sont dus à une confusion quant aux rôles ou à des lacunes dans les attentes. Lorsque les groupes d'ingénierie, de réglementation et de fabrication apportent leur expertise de manière synchronisée, la validation devient bien plus efficace et fiable.
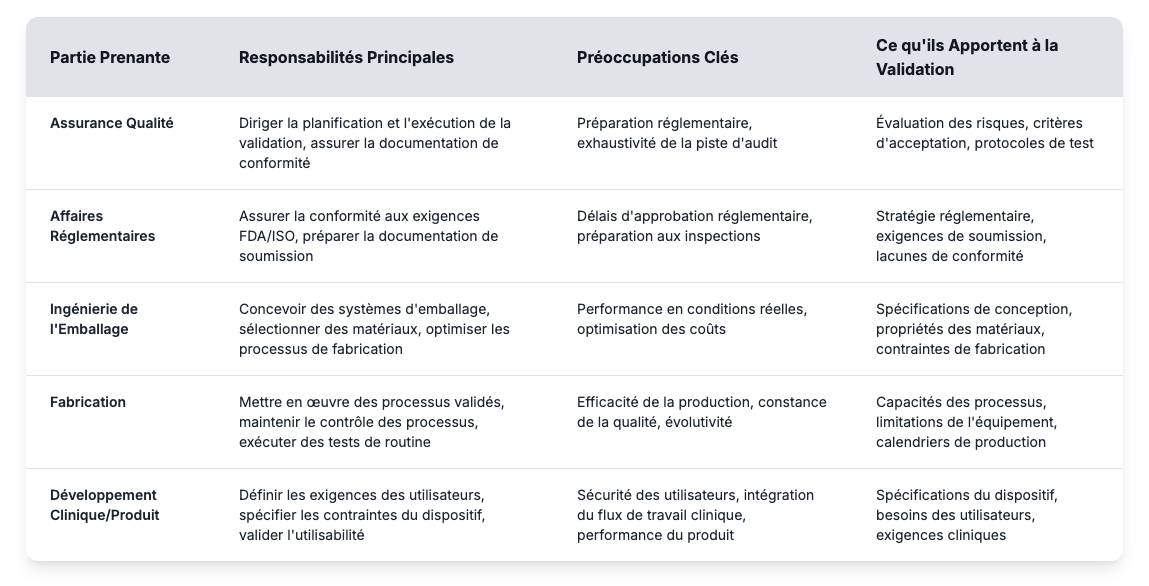
La définition de ces rôles permet aux fabricants de dispositifs médicaux de mettre en place des programmes de validation efficaces et d'éviter les pièges courants. L'évaluation des risques permet d'identifier les points de défaillance potentiels tandis que les critères d'acceptation fixent des objectifs mesurables pour les performances des emballages. Les protocoles de test définissent comment les tests de validation permettront de prouver que les systèmes de barrières stériles répondent aux exigences réglementaires pendant toute la durée de conservation prévue. L'élaboration d'un programme de validation efficace commence par la compréhension de ces exigences fondamentales et la mise en place de processus clairs qui relient les contributions des parties prenantes à des résultats mesurables.
Exigences fondamentales pour la validation des emballages
Tout programme de validation réussi commence par trois éléments fondamentaux de validation des emballages que les régulateurs s'attendent à voir :
- Plan de validation complet : Votre plan de validation relie les décisions de conception à la documentation de conformité. Il définit qui fait quoi, quand les tests ont lieu et comment vous allez documenter les résultats. Les régulateurs examinent ces plans lors des inspections pour voir si votre approche est logique ou si vous ne faites que cocher des cases. Les bons plans comprennent des protocoles clairs avec des attributions de responsabilités ainsi que des délais réalistes qui tiennent compte des retards potentiels.
- Évaluation structurée des risques : L'évaluation des risques vous permet de concentrer les ressources de validation là où elles sont le plus importantes. Vous identifierez des scénarios dans lesquels l'emballage pourrait compromettre la stérilité ou la sécurité de l'appareil, par exemple des défaillances du scellage lors de l'expédition, des problèmes matériels liés à votre méthode de stérilisation ou un emballage trop difficile à ouvrir pour les utilisateurs en toute sécurité. Cette évaluation vous indique quels tests sont essentiels et lesquels sont intéressants.
- Critères d'acceptation scientifiquement justifiés : Les critères d'acceptation définissent ce que signifie « suffisamment bon » pour les performances de votre emballage. Ils doivent être spécifiques et mesurables tout en restant liés à la sécurité des patients plutôt qu'à la commodité de fabrication. Au lieu de définir des critères vagues tels que « apparence acceptable », vous allez fixer des objectifs de résistance de l'étanchéité, de taux de fuite et de maintien de la stérilité adaptés à votre appareil et aux conditions d'utilisation spécifiques.
La faiblesse des bases crée des problèmes de validation qui se multiplient à mesure que les programmes progressent. Les équipes découvrent souvent des lacunes trop tard, alors que les corriger implique de recommencer à zéro avec des tests qui auraient dû être inclus dès le début. L'effet domino est prévisible : la dispersion des tests conduit à des preuves incomplètes, ce qui soulève des questions réglementaires qui retardent les approbations et augmentent les coûts.
Les quatre piliers de la validation des emballages
La norme ISO 11607 divise la validation des emballages en quatre piliers interconnectés qui fonctionnent ensemble pour prouver que votre emballage ne tombera pas en panne au moment le plus important. Plutôt que de les traiter comme des cases à cocher distinctes, les équipes performantes comprennent comment chaque pilier alimente le suivant pour établir des preuves complètes que l'emballage protège la stérilité des appareils tout au long de leur cycle de vie.
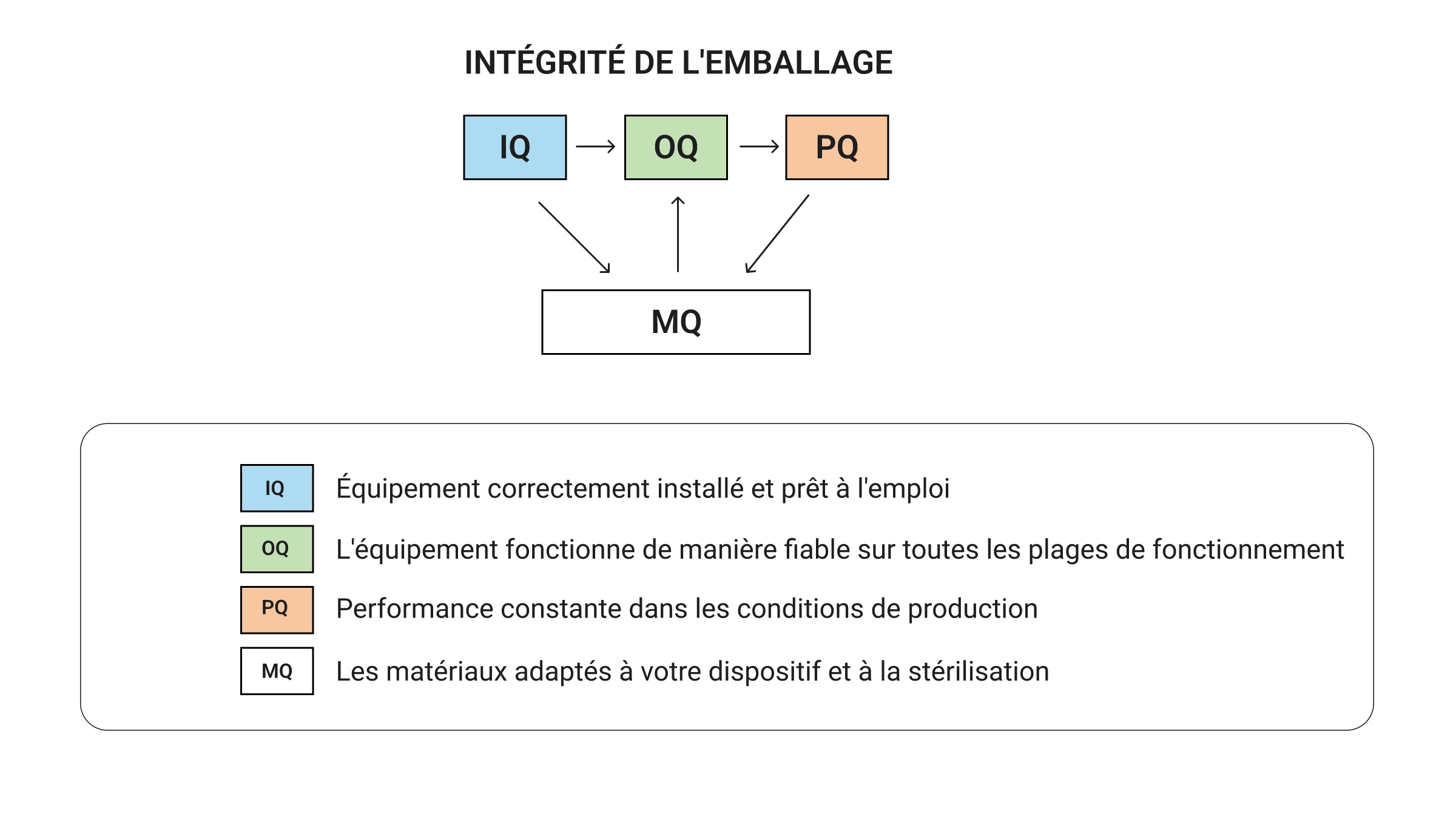
Vous ne pouvez pas prouver la constance des performances d'emballage (PQ) sans savoir que votre équipement fonctionne de manière fiable (OQ). Vous ne pouvez pas vous fier aux performances d'un équipement sans une installation correcte (IQ). La qualification des matériaux (MQ) va de pair avec la validation de l'équipement pour garantir que tout fonctionne ensemble en tant que système d'emballage complet. Lorsqu'elle est mise en œuvre correctement, cette approche systématique permet d'obtenir des preuves de validation étape par étape, chaque pilier renforçant les autres. Lorsque vous sautez des étapes, vous créez des problèmes coûteux :
- Si vous ignorez le QI, vous ne savez pas si des problèmes d'équipement faussent les résultats de vos tests ultérieurs.
- Un QO faible vous laisse avec des paramètres de processus qui semblent satisfaisants lors des tests, mais qui s'effondrent en raison des contraintes de production.
- Le raccourcissement de la qualification des matériaux est la façon dont les équipes se retrouvent avec des emballages qui échouent lors de l'expédition ou perdent leur stérilité au fil du temps.
Pensez à la validation comme à la construction d'une maison : vous ne commenceriez pas par le toit avant d'avoir posé des fondations solides. Les quatre piliers fonctionnent de la même manière, chaque étape soutenant la suivante. Oubliez le travail préparatoire et vous consacrerez beaucoup plus de temps et d'argent à la résolution de problèmes qui auraient pu être évités grâce à une approche systématique dès le départ.
Qualification de l'installation : vérification et étalonnage de l'équipement
Sans documentation de base solide, les problèmes d'équipement et les défaillances des processus deviennent impossibles à distinguer lors des étapes de validation ultérieures. La qualification de l'installation (IQ) permet d'éviter cela en établissant exactement par quoi vous commencez avant le début de tout test de performance.
Le principal produit d'IQ est la documentation des paramètres de base qui capture le point de départ de votre équipement. Vous allez enregistrer :
- Plages de fonctionnement pour des paramètres critiques tels que la température, la pression, la vitesse et le temps de séjour
- Paramètres par défaut et configurations du système de contrôle
- Conditions environnementales dans la zone d'emballage
- Caractéristiques de performance de l'équipement à vide
- Exigences et programmes de maintenance préventive qui permettent à l'équipement de fonctionner de manière constante
La vérification et l'étalonnage de l'équipement constituent la base de ces bases de référence. Vous confirmerez que tous les composants correspondent aux spécifications d'achat, que les services publics répondent aux exigences et que les systèmes de sécurité fonctionnent correctement. Pour les équipements de scellage, vous documenterez les capacités de température et de pression ainsi que les spécifications des éléments chauffants. Les systèmes de contrôle sont vérifiés pour confirmer qu'ils peuvent maintenir les paramètres requis.
L'étalonnage étend les bases de l'IQ. Les instruments qui surveillent ou contrôlent le processus d'emballage (régulateurs de température, manomètres, systèmes de chronométrage et outils de test) doivent disposer de certificats traçables selon les normes nationales. Ces enregistrements ancrent les performances de l'équipement et démontrent le contrôle des processus lors de la validation et de la production de routine.
Ce record devient également le point de référence pour les étapes suivantes. Si la résistance du joint varie pendant la qualification des performances, les résultats peuvent être attribués à la variation de l'équipement documentée dans IQ. Sans ce niveau de documentation, il devient difficile de faire la distinction entre les problèmes d'équipement et les défaillances des processus, ce qui entraîne des enquêtes coûteuses qu'un travail initial approfondi aurait pu éviter.
Qualification opérationnelle : essais sur toutes les plages de fonctionnement
Votre équipement peut fonctionner parfaitement dans des conditions idéales, mais donner des résultats incohérents lorsque la production devient compliquée. La qualification opérationnelle (OQ) révèle ces zones problématiques avant qu'elles ne causent des problèmes en aval. En testant les paramètres à leurs limites, les équipes peuvent voir où le processus se maintient et où les performances commencent à se dégrader.
Votre objectif est d'établir des paramètres de processus que les équipes de production utiliseront quotidiennement : les limites de fonctionnement avec des points de consigne normaux ainsi que des plages de variation acceptables. Vous trouverez des gammes où l'équipement produit des résultats fiables de manière constante, avec des réglages optimaux qui offrent les meilleures performances et des marges de sécurité qui permettent d'éviter les problèmes. Pour tester la fonctionnalité de l'équipement, il faut faire varier systématiquement un paramètre tout en maintenant les autres constants, puis surveiller la qualité des résultats des emballages. Les tests en périphérie permettent d'identifier où les performances commencent à se dégrader. Cela devient votre marge de sécurité pour la production de routine.
Les dossiers OQ doivent aller au-delà de la liste de ce que l'équipement peut faire. Ils devraient également saisir les conditions qui permettent de maintenir la cohérence des résultats. Par exemple, une humidité élevée peut nuire à l'étanchéité, les changements de température peuvent entraîner une variation des paramètres critiques et les machines situées à proximité peuvent introduire des vibrations qui réduisent la précision.
Ces tests détectent les combinaisons de paramètres susceptibles de créer des problèmes en production avant qu'ils n'apparaissent réellement. Votre équipement peut fonctionner à certains réglages mais donner des résultats incohérents, ce qui peut entraîner des maux de tête en aval. OQ identifie ces zones problématiques afin que les équipes de production puissent s'en sortir. Les plages que vous définissez ici constituent la base de la formation des opérateurs et des alarmes de processus qui assurent le bon fonctionnement de la fabrication.
Qualification des performances : preuve de performances d'emballage constantes
La qualification de performance (PQ) démontre que l'ensemble de votre processus d'emballage produit systématiquement des emballages qui répondent à tous les critères d'acceptation dans des conditions de production réelles. C'est ici que la validation passe de la preuve de la capacité à la preuve de la fiabilité.
La réalité est que votre processus d'emballage doit fonctionner lorsque tout n'est pas parfait. Les réglages de l'équipement varient légèrement, les opérateurs changent entre les équipes et la qualité des lots de matériaux varie. Les conditions environnementales fluctuent tout au long de la journée. Les tests PQ montrent que votre procédé produit toujours des emballages acceptables malgré ces variations normales. Les tests dans le pire des cas vont encore plus loin en poussant votre processus validé à des limites réalistes. Les tests de résistance devraient refléter les réalités de la production, et pas seulement les conditions idéales. Cela peut impliquer de fonctionner à des vitesses de ligne maximales pendant que les opérateurs font pression pour respecter les délais, de travailler dans des conditions environnementales réduites si les systèmes CVC ne fonctionnent pas bien, ou de tester la qualité à la fin d'un quart de travail lorsque la fatigue peut influencer les résultats.
Pour effectuer des tests de validation dans des conditions de production réelles, il faut utiliser les mêmes matériaux, les mêmes paramètres d'équipement et les mêmes conditions environnementales que ceux auxquels les équipes de fabrication sont confrontées quotidiennement. Il est plus important de saisir les variations des processus naturels au fil du temps, des opérateurs, des lots de matériaux et de l'état des équipements que de tester dans des conditions contrôlées artificiellement qui ne sont pas représentatives de la réalité. La planification statistique définit le nombre de tests nécessaires pour prouver des performances constantes. Un plan d'échantillonnage bien conçu couvre plusieurs cycles et opérateurs, garantissant ainsi que les résultats reflètent la capacité réelle du procédé et non le hasard. Ces preuves orientent ensuite la surveillance des processus et garantissent des performances validées au cours de la fabrication quotidienne.
Qualification des matériaux et de la conception : garantir la compatibilité et la protection
La qualification des matériaux (MQ) montre que les matériaux d'emballage des appareils peuvent fonctionner comme prévu tout au long de leur cycle de vie. Pour les dispositifs médicaux stérilisés au stade terminal, cela signifie qu'il faut sélectionner un emballage primaire qui résiste à la méthode de stérilisation choisie et qui continue de protéger la stérilité pendant la distribution et le stockage. La validation du système d'emballage confirme ensuite que les matériaux, la conception et le scellage fonctionnent ensemble comme une barrière stérile pendant toute la durée de vie prévue de l'appareil.
Lors de la sélection des matériaux, les équipes doivent évaluer trois facteurs :
- La méthode de stérilisation qui sera utilisée
- L'environnement de stockage auquel le colis doit résister
- Les exigences des utilisateurs pour une ouverture et un accès sécurisés à l'appareil
Chaque méthode de stérilisation présente des défis uniques. La stérilisation à l'oxyde d'éthylène nécessite généralement des matériaux qui résistent à la dégradation chimique tout en permettant la pénétration et l'évacuation des gaz pour une stérilisation et une aération efficaces. La stérilisation aux rayons gamma expose les emballages à des radiations susceptibles d'en affaiblir la résistance et la flexibilité. La stérilisation à la vapeur associe une température élevée à une humidité susceptible de compromettre les propriétés de barrière. Si ces risques ne sont pas traités rapidement, la barrière stérile peut échouer en cas d'utilisation réelle, mettant en danger à la fois la conformité et la sécurité des patients.
Les tests de compatibilité des matériaux évaluent la manière dont votre emballage interagit avec les composants de l'appareil pendant toute la durée de conservation prévue, mais ils doivent également tenir compte de la facilité d'utilisation pratique. Les matériaux doivent également conserver leurs propriétés de barrière microbienne pour garantir que les barrières stériles continuent de bloquer les contaminants pendant toute la durée de conservation prévue. Dans le même temps, la résistance au pelage doit être suffisamment forte pour maintenir l'intégrité du joint pendant la distribution, mais suffisamment douce pour que les professionnels de santé puissent l'ouvrir de manière aseptique sans se déchirer. Vous réaliserez des études de vieillissement accéléré qui simuleront des années de stockage en quelques semaines, combinées à un vieillissement en temps réel dans des conditions de stockage réelles pour confirmer la validité de vos prévisions.
Protocoles de test des matériaux :
Les protocoles de test initiaux permettent d'établir les performances de référence des matériaux avant le début des études de validation complètes. Concentrez-vous sur la conception de protocoles de test qui sollicitent les matériaux de la même manière qu'ils le seront réellement dans le monde réel, plutôt que de répondre à des spécifications de test arbitraires qui ne sont pas liées aux exigences de performance réelles. Les caractéristiques d'ouverture doivent permettre un accès propre et contrôlé qui ne compromet pas la stérilité et ne diffuse pas de particules dans le champ stérile, ce qui signifie que vos tests doivent simuler des conditions d'utilisation cliniques réelles plutôt que des environnements de laboratoire idéaux.
Tests de distribution et de transit : simulation du transport maritime dans le monde réel
L'emballage doit résister à tout le trajet entre l'atelier de fabrication et le chevet du patient. Les tests de distribution recréent les contraintes liées à l'expédition et à la manutention afin que les équipes puissent confirmer que les barrières stériles restent intactes et fonctionnelles tout au long de la chaîne d'approvisionnement.
ASTM D4169 et Protocoles ISTA expliquez comment simuler des environnements de distribution, mais vous devez les adapter à vos conditions d'expédition réelles. Au lieu de procéder à des contrôles isolés, les tests devraient porter sur l'ensemble du trajet : un colis peut tomber, être secoué par les vibrations du transport ou écrasé sous le chargement de l'entrepôt. En cours de route, les changements d'orientation, les manipulations répétées et les conditions climatiques changeantes mettent à l'épreuve la résistance de la barrière stérile. L'exécution de ces contraintes en séquence donne une image plus fidèle des performances de l'emballage dans le monde réel de la distribution.
Les packages sont soumis à plusieurs contrôles après les tests de distribution. L'inspection visuelle permet de détecter les dommages à la surface, tandis que les tests de résistance confirment que les fermetures restent intactes. Des méthodes plus sensibles, telles que les tests à bulles, permettent de détecter des microfuites invisibles à l'œil nu dans des barrières stériles. La dernière étape consiste à effectuer des tests d'intégrité, qui prouvent que la barrière microbienne fonctionne toujours même après une simulation d'expédition. Ces résultats n'ont d'importance que si les tests reflètent les conditions d'expédition réelles. Les colis qui respectent des protocoles génériques mais échouent sur le terrain entraînent des rappels, ce qui nuit à la réputation de l'entreprise. Plus important encore, ils mettent les patients en danger.

Études sur le vieillissement accéléré et en temps réel : prouver les allégations relatives à la durée de conservation
Les allégations relatives à la durée de conservation sont maintenues ou diminuées lorsqu'il est prouvé que l'emballage préserve sa barrière stérile aussi longtemps que les dispositifs restent sur le marché. Le vieillissement accéléré donne aux fabricants une vision précoce des performances des emballages en utilisant une température et une humidité élevées pour simuler des années de stockage en quelques semaines seulement. NORME ASTM F 1980 (version actuelle) décrit l'approche, mais la vraie difficulté est de concevoir des conditions qui reflètent les environnements réels sans introduire des modes de défaillance qui ne se produiraient jamais dans la pratique. Lorsqu'elles sont bien conçues, ces études produisent des données préliminaires qui appuient les allégations relatives à la durée de conservation initiale et aident les produits à aller de l'avant alors que des études à plus long terme sont toujours en cours.
Le vieillissement en temps réel complète le tableau. Les emballages issus des derniers cycles de production sont stockés dans des conditions réelles et leurs performances sont contrôlées pendant des mois, voire des années. Ces preuves montrent comment les emballages résistent aux réalités quotidiennes du stockage et de la distribution, fournissant les données de confirmation attendues par les régulateurs. Les équipes qui commencent tôt les études en temps réel évitent les lacunes dans les données qui peuvent ralentir les approbations ou les renouvellements ultérieurs.
Les déterminations de durée de conservation les plus fiables proviennent de la combinaison de projections accélérées et de confirmations en temps réel. L'analyse statistique indique à quel moment les propriétés de l'emballage sont inférieures à vos objectifs et vous donne des intervalles de confiance pour les allégations relatives à la durée de conservation. Une mauvaise planification peut toutefois saper l'ensemble de l'effort : tester des prototypes plutôt que des cycles de production, sélectionner des conditions de stockage qui ne reflètent pas la réalité ou sous-estimer la taille des échantillons sont autant de facteurs qui nuisent à la crédibilité.
Avec la bonne approche, les études sur le vieillissement prouvent que les emballages protégeront les patients pendant toute la durée de vie prévue, tout en donnant aux fabricants et aux régulateurs la certitude que les allégations seront maintenues au fil du temps. Ces preuves vont au-delà des exigences réglementaires pour démontrer des performances réelles qui sont importantes pour la sécurité des patients.
Évaluation de l'utilisabilité et facteurs humains : validation de l'interaction utilisateur
UNE système d'emballage peut répondre à toutes les normes techniques et échouer s'il ne peut pas être ouvert en toute sécurité entre les mains d'un clinicien. La validation de l'utilisabilité garantit que les systèmes de barrière stérile résistent non seulement au laboratoire, mais également au moment réel où les dispositifs sont ouverts pour les soins aux patients. La FDA et la norme ISO 11607 lient directement l'utilisabilité à la gestion des risques, reconnaissant qu'un emballage qui compromet la technique d'asepsie est en fait un système défaillant.
L'un des principaux objectifs est test de présentation aseptique - la preuve que la barrière stérile peut être ouverte en toute sécurité dans la pratique. Les systèmes de fermeture et les scellés des emballages doivent résister à une défaillance prématurée tout en s'ouvrant facilement dans des conditions cliniques. L'intégrité des joints est essentielle. Les emballages doivent être pelés proprement sans se déchirer, et la conception doit empêcher les particules ou les débris de pénétrer dans le champ stérile. Un problème fréquent est le joint surdimensionné, suffisamment solide pour atteindre les objectifs de test mais si difficile à ouvrir qu'une technique aseptique devient impraticable. Les tests d'utilisation simulés permettent de détecter ces problèmes avant que les produits ne soient mis sur le marché.
Dans la pratique, les tests impliquent souvent que des infirmières ou des techniciens chirurgicaux ouvrent les emballages dans des conditions stériles pendant que les observateurs remarquent toute rupture technique. Le type de gant est important : le nitrile et le latex offrent une adhérence et une sensation différentes. D'autres facteurs du monde réel, tels que l'éclairage de la salle d'opération, la pression temporelle pendant les procédures ou la capacité de coopération de l'emballage lui-même, affectent également la capacité à maintenir la stérilité. Même des détails tels que les languettes amovibles peuvent faire la différence : facile à manipuler à mains nues, mais presque impossible avec des gants. Une résistance d'étanchéité excessive présente le même risque, obligeant à trouver des solutions qui vont à l'encontre de l'objectif même de l'emballage stérile. Un sceau qui ne peut pas être ouvert de manière aseptique ne fait pas que frustrer les utilisateurs, il compromet directement la stérilité sur le lieu de soin.
Il est tout aussi important de comprendre comment les personnes interagissent avec le package dans la pratique. Des études d'utilisabilité montrent que des professionnels de santé ouvrent des appareils dans des conditions réalistes, faisant apparaître des moments d'hésitation ainsi que des variations techniques ou un risque de contamination potentiel. Ces observations révèlent des problèmes qui échappent aux tests sur les matériaux : des joints qui exigent une force excessive ou des emballages qui se déchirent de façon imprévisible lorsque les utilisateurs ont besoin d'un accès rapide. Des indices d'ouverture peu clairs créent une confusion qui compromet la technique stérile pendant les procédures réelles.
Enfin, la validation doit tenir compte de exigences en matière de formation. Si une utilisation sûre dépend d'instructions complexes, le risque d'erreur augmente. L'évaluation précoce des besoins de formation permet de confirmer que l'emballage est intuitif pour les utilisateurs auxquels il est destiné. Toutes les instructions essentielles pour une manipulation en toute sécurité doivent être clairement documentées. Les équipes qui prennent cette mesure dès le départ sont mieux placées pour éviter des refontes coûteuses ou des plaintes inattendues après le lancement.
Lorsque l'utilisabilité est intégrée à la validation dès le départ, les fabricants obtiennent bien plus que des preuves de conformité. Ils proposent des emballages qui conviennent aussi bien aux patients qu'aux cliniciens, tout en garantissant aux régulateurs qu'il fonctionne là où cela compte le plus.
Qualification des performances des procédés : validation de la cohérence de la fabrication
Même l'emballage le plus soigneusement conçu échoue si le processus de fabrication ne permet pas d'obtenir le même résultat à chaque fois. La qualification des performances des processus (PPQ) prouve que les cycles d'emballage ne se contentent pas de réussir en laboratoire ou lors des essais de validation, mais qu'ils restent cohérents à pleine échelle de production. C'est à ce stade que les régulateurs et les équipes de qualité internes ont besoin de preuves que les systèmes d'emballage peuvent résister à une production réelle.
Un PPQ solide examine l'ensemble de la séquence de production, y compris les processus d'assemblage. Il examine la manière dont les matériaux sont stockés, la manière dont les opérateurs manipulent les composants et si les contrôles environnementaux permettent de maintenir la stabilité des conditions. Toute faiblesse dans ces domaines peut éroder les performances de l'emballage, même lorsque les paramètres de l'équipement sont modifiés.
La fiabilité vient de la répétabilité. C'est pourquoi le PPQ nécessite plusieurs cycles de production pour prouver que le processus fournit des résultats cohérents sur les différents lots. Les équipes valident généralement trois lots consécutifs pour montrer que les performances sont constantes d'un cycle de production à l'autre, bien que les exigences puissent varier en fonction des risques liés au produit et des directives réglementaires. Un suivi continu permet ensuite de maintenir cette assurance, en détectant les dérives avant qu'elles ne se transforment en problèmes de qualité.
Les avantages vont bien au-delà de la conformité. Un PPQ bien exécuté offre des avantages qui se répercutent largement sur les opérations quotidiennes. Elle permet de rendre la production plus prévisible tout en accélérant les décisions de lancement. Plus important encore, cela renforce la confiance des régulateurs dans le fait que le système est sous contrôle.
Exigences de revalidation et gestion du changement
Le changement est inévitable dans le secteur de la fabrication, et chaque changement soulève la question de savoir si votre système d'emballage fait toujours son travail. La revalidation fournit les preuves que c'est le cas, empêchant ainsi de petits ajustements de créer des risques importants. Les régulateurs considèrent les changements incontrôlés comme un signal d'alarme, et les fabricants devraient également le faire. Les déclencheurs courants sont les suivants :
- Mises à jour de l'équipement, telles que de nouveaux systèmes d'étanchéité ou d'inspection
- Modifications de la stérilisation, qu'il s'agisse d'un changement de méthode ou d'ajustements de paramètres
- Modifications des matériaux, du changement de fournisseur à la sélection de différents substrats
- Changements de processus, même mineurs, qui modifient les conditions de fonctionnement
L'étendue de la revalidation dépend du risque et des exigences de validation documentées. Toute modification d'un procédé soulève la question de la stérilité. Les équipes peuvent constater qu'un petit ajustement affecte l'intégrité du joint. Dans d'autres cas, la compatibilité des matériaux doit être réévaluée. Parfois, la question est de savoir si les propriétés de barrière microbienne seront maintenues. Quel que soit le déclencheur, les tests de suivi fournissent la preuve que l'emballage continue à protéger comme prévu. Des examens réguliers sont également attendus, car l'équipement s'use avec le temps. Les changements de fournisseurs ou la dérive progressive des processus peuvent avoir le même impact, et chacun d'entre eux doit être surveillé.
Une bonne documentation réunit tous ces éléments pour en faire des preuves auxquelles les régulateurs peuvent avoir confiance. Il doit expliquer le raisonnement qui sous-tend chaque test, et pas simplement indiquer que le test a eu lieu. Des dossiers clairs fournissent bien plus qu'une simple trace écrite. Ils donnent confiance aux régulateurs, aident les équipes à gérer le changement de manière proactive et, surtout, montrent que les emballages continuent de protéger les patients bien au-delà du premier rapport de validation.
Défis courants et solutions efficaces
La validation peut échouer pour des raisons bien connues. La taille des échantillons est souvent sous-estimée, les tests dans le pire des cas sont ignorés et les fiches documentaires sont conservées jusqu'à ce qu'elles ne reflètent plus ce qui a été fait. Les équipes passent également des heures à comparer manuellement les documents et à se demander si elles ont détecté tous les changements critiques. Chacun de ces faux pas peut sembler insignifiant sur le moment, mais ensemble, ils entraînent des retouches coûteuses et suscitent des conclusions réglementaires qui nuisent à la crédibilité longtemps après la fin de l'audit. Le goulot d'étranglement lié à l'évaluation manuelle est particulièrement problématique car il oblige les professionnels de la qualité expérimentés à effectuer des comparaisons fastidieuses au lieu d'appliquer leur expertise à l'évaluation des risques et à la prise de décisions stratégiques.
L'approche la plus efficace consiste à faire de la validation une activité stratégique. Des critères d'acceptation clairs définis dès le départ permettent d'éviter les désaccords à un stade avancé, en particulier lorsque les équipes chargées de la qualité, de l'ingénierie et de la réglementation s'alignent dès le début du processus. Lorsque la validation se déroule tout au long du développement au lieu d'être traitée comme une étape finale, les lacunes deviennent plus faciles à repérer tandis que les examens réglementaires se déroulent plus facilement.
Pour travailler efficacement, il faut concentrer les efforts là où cela compte le plus, et pas simplement faire des économies. L'échantillonnage basé sur les risques se concentre sur les ressources où l'échec serait le plus important. Les plans de test peuvent être conçus pour répondre à plusieurs exigences à la fois, réduisant ainsi la redondance sans nuire à la rigueur. Utilisées de cette façon, les meilleures pratiques permettent de gagner du temps tout en renforçant la base de preuves, en garantissant que l'emballage gagne la confiance des régulateurs et continue de protéger les patients qui l'utilisent.
Documentation et soumission réglementaire
Pour les régulateurs, si cela n'est pas documenté, cela ne s'est pas produit. Les fabricants sont confrontés à la même réalité : des dossiers incomplets peuvent ralentir les approbations tout en déclenchant des résultats d'inspection qui affaiblissent la confiance dans le système qualité.
Une documentation solide permet de combler ces lacunes. Un enregistrement complet montre comment les tests ont été planifiés et exécutés tout en reliant les résultats aux critères d'acceptation. Cette documentation atteste de la conformité réglementaire. Les certificats de validation et les dossiers techniques formalisent ensuite ces preuves, confirmant que les systèmes d'emballage répondent à la fois aux attentes de la norme ISO 11607 et de la FDA. Plus qu'un simple classeur de rapports, une documentation efficace associe des protocoles, des données et des évaluations des risques dans un récit de conformité contrôlé et prêt à être inspecté. Lorsque les régulateurs le demandent, les fabricants doivent montrer non seulement ce qui a été testé, mais aussi pourquoi, comment et avec quels résultats.
Une documentation complète ne satisfait pas seulement les régulateurs ; elle permet également de garantir la mise sur le marché des appareils sans interruptions coûteuses. Lorsque les dossiers sont complets et prêts à être inspectés, les fabricants peuvent défendre leur processus en toute confiance et les patients reçoivent un emballage qui les protège tout au long du cycle de vie de l'appareil.
Garantir le succès du système d'emballage
La validation des emballages prouve que les systèmes de barrières stériles peuvent supporter le trajet entre l'usine et le point de service. Basée sur une planification minutieuse et étayée par des tests et une documentation solides, la validation donne aux fabricants l'assurance que leurs emballages seront performants sur le terrain et satisferont les régulateurs. Traiter la validation comme une responsabilité du cycle de vie, et non comme un exercice ponctuel, permet d'éviter des échecs coûteux et de renforcer la confiance au sein des équipes de qualité et des régulateurs.
Le paysage évolue. Les plateformes de validation numérique et les liens plus étroits avec la gestion des risques redéfinissent les meilleures pratiques, tandis que les régulateurs s'orientent vers un plus grand alignement mondial. Ce qui ne changera pas, c'est l'objectif : veiller à ce que les emballages restent stériles afin que les cliniciens puissent compter sur chaque appareil et que les patients bénéficient de la protection qu'ils méritent. À l'avenir, l'intégration plus étroite de la validation des emballages et de la validation de la stérilisation façonnera les pratiques de l'industrie, garantissant ainsi la prise en charge complète des allégations de stérilité.
La vérification de GlobalVision accélère la validation des emballages en automatisant l'inspection des documents et des illustrations, ce qui permet aux équipes de se conformer en toute confiance sans les ralentir.
