
Wenn ein steriles Medizinprodukt bei einer Operation versagt, weil seine Verpackung die Sterilität während des Transports nicht aufrechterhalten konnte, leidet zuerst die Patientensicherheit. Produktrückrufe und ein beschädigter Ruf des Unternehmens folgen dicht darauf.
Einführung in die Verpackungsvalidierung
Die Validierung der Verpackung von Medizinprodukten beweist, dass Ihre Verpackungssysteme die Sterilität der Geräte von der Herstellung bis hin zum Point-of-Care-Prozess schützen. Ihre Verpackung dient als steriles Barrieresystem, das den mikrobiellen Schutz während der gesamten Sterilisation und Verteilung gewährleistet, sodass Geräte jahrelang steril bleiben, bevor sie die Patienten erreichen.
ISO 11607 bietet den internationalen Rahmen für die Validierung dieser Systeme. Teil 1 legt Anforderungen an Materialien und sterile Barrieresysteme fest, die zur Verpackung endsterilisierter Medizinprodukte verwendet werden. Teil 2 enthält die Validierungsanforderungen für Prozesse in den Bereichen Formung, Versiegelung und Sterilbarriere. Zusammen stellen sie sicher, dass die Verpackung sowohl in der Lage ist, die Sterilität aufrechtzuerhalten, als auch unter realen Herstellungsbedingungen ihre Leistung bewiesen hat. FDA-Verordnung zum Qualitätssystem (21 CFR 820) setzt parallele Anforderungen durch Entwurfskontrollen und Prozessvalidierungsmandate durch, sodass die Validierung eher zu einer Konformitätsvoraussetzung als zu einer optionalen Qualitätsmaßnahme wird.
Die Validierung wird in der Regel von der Qualitätssicherung geleitet, doch der tatsächliche Erfolg hängt von der abteilungsübergreifenden Koordination ab. Viele Verzögerungen sind auf Unklarheiten über die Rollen oder auf Lücken in den Erwartungen zurückzuführen. Wenn Konstruktions-, Regulierungs- und Fertigungsgruppen ihr Fachwissen synchron einbringen, wird die Validierung weitaus effizienter und zuverlässiger.

Die richtigen Rollen helfen Herstellern von Medizinprodukten dabei, effektive Validierungsprogramme zu entwickeln und häufige Fallstricke zu vermeiden. Bei der Risikobewertung werden potenzielle Fehlerquellen identifiziert, während die Akzeptanzkriterien messbare Ziele für die Verpackungsleistung festlegen. In den Testprotokollen ist festgelegt, wie durch Validierungstests nachgewiesen wird, dass sterile Barrieresysteme die gesetzlichen Anforderungen während der gesamten vorgesehenen Haltbarkeitsdauer erfüllen. Der Aufbau eines erfolgreichen Validierungsprogramms beginnt mit dem Verständnis dieser grundlegenden Anforderungen und der Etablierung klarer Prozesse, die die Beiträge der Interessengruppen mit messbaren Ergebnissen verbinden.
Grundlegende Anforderungen für die Verpackungsvalidierung
Jedes erfolgreiche Validierungsprogramm beginnt mit drei grundlegenden Elementen der Verpackungsvalidierung, die die Aufsichtsbehörden erwarten:
- Umfassender Validierungsplan: Ihr Validierungsplan verbindet Konstruktionsentscheidungen mit der Konformitätsdokumentation. Er definiert, wer was tut, wann die Tests stattfinden und wie Sie die Ergebnisse dokumentieren. Die Aufsichtsbehörden prüfen diese Pläne bei Inspektionen, um festzustellen, ob Ihr Ansatz sinnvoll ist oder ob Sie nur die Kästchen ankreuzen. Gute Pläne beinhalten klare Protokolle mit Zuständigkeitszuweisungen sowie realistische Zeitpläne, die mögliche Verzögerungen berücksichtigen.
- Strukturierte Risikobewertung: Die Risikobewertung hilft Ihnen dabei, die Validierungsressourcen dort zu konzentrieren, wo sie am wichtigsten sind. Sie identifizieren Szenarien, in denen die Verpackung die Sterilität oder Sicherheit des Geräts gefährden könnte — Dinge wie Versagen der Versiegelung während des Transports, Materialprobleme bei Ihrer Sterilisationsmethode oder Verpackungen, die für Benutzer zu schwer zu öffnen sind. Anhand dieser Bewertung erfahren Sie, welche Tests entscheidend sind und welche sinnvoll sind.
- Wissenschaftlich begründete Aufnahmekriterien: Akzeptanzkriterien definieren, wie „gut genug“ für Ihre Verpackungsleistung aussieht. Sie müssen spezifisch und messbar sein und gleichzeitig der Patientensicherheit und nicht der Bequemlichkeit der Herstellung dienen. Anstatt vage Kriterien wie „akzeptables Aussehen“ festzulegen, legen Sie Ziele für Dichtheit, Leckrate und Aufrechterhaltung der Sterilität fest, die für Ihr spezifisches Gerät und Ihre Einsatzbedingungen sinnvoll sind.
Schwache Grundlagen führen zu Validierungsproblemen, die sich mit dem Fortschreiten der Programme vervielfachen. Teams entdecken Lücken oft zu spät, wenn deren Behebung bedeutet, mit Tests, die von Anfang an hätten enthalten sein sollen, von vorne zu beginnen. Der Dominoeffekt ist vorhersehbar: Vereinzelte Tests führen zu unvollständigen Nachweisen, was zu regulatorischen Fragen führt, die Genehmigungen verzögern und die Kosten erhöhen.
Die vier Säulen der Verpackungsvalidierung
ISO 11607 unterteilt die Verpackungsvalidierung in vier Teile miteinander verbundene Säulen die zusammenarbeiten, um zu beweisen, dass Ihre Verpackung nicht versagt, wenn es darauf ankommt. Anstatt diese als separate Kontrollkästchen zu behandeln, verstehen erfolgreiche Teams, wie jede Säule in die nächste übergeht, um umfassende Beweise dafür zu liefern, dass Verpackungen die Sterilität der Geräte während des gesamten Lebenszyklus des Geräts schützen.
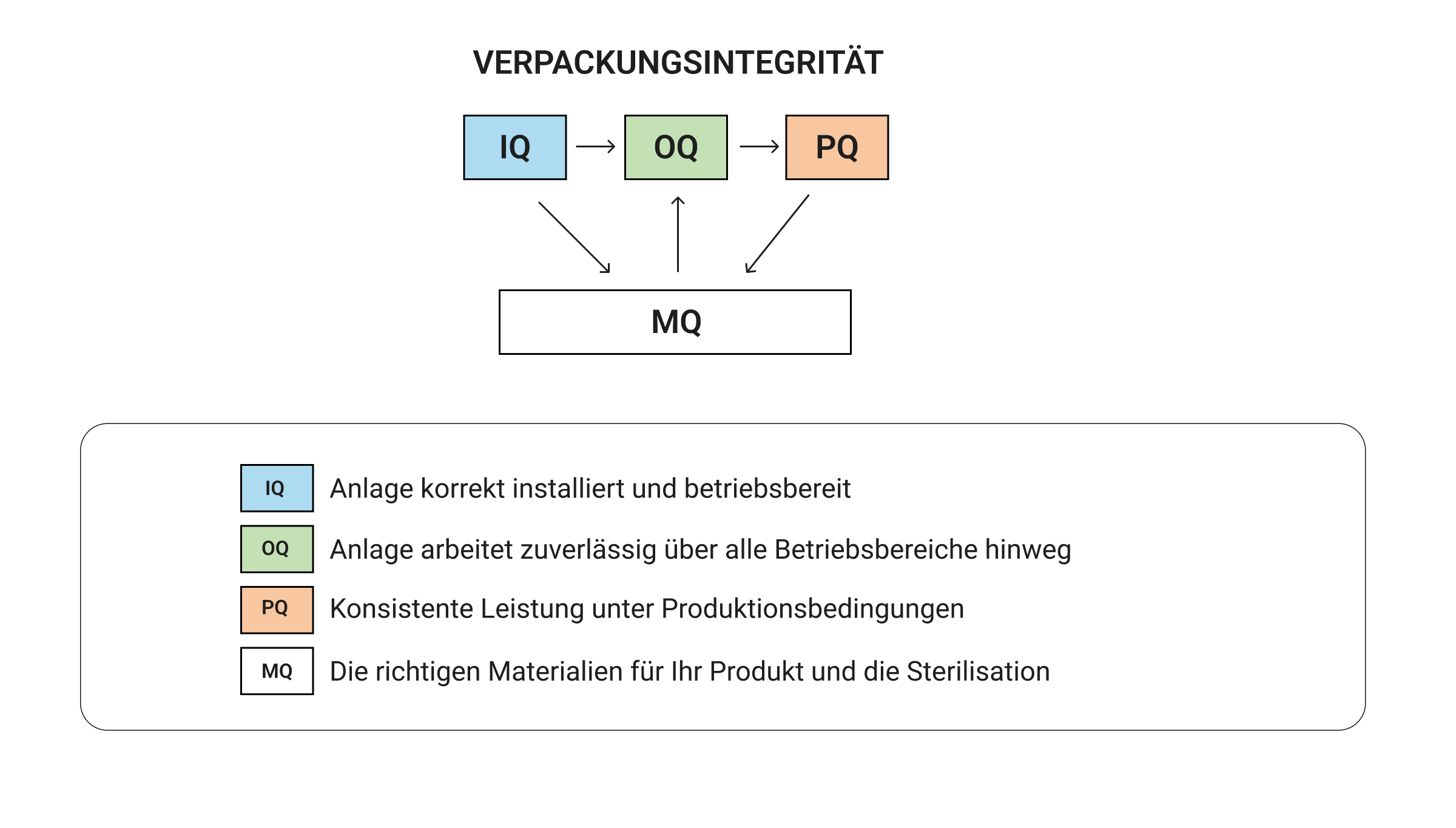
Sie können keine gleichbleibende Verpackungsleistung (PQ) nachweisen, ohne zu wissen, dass Ihre Geräte zuverlässig funktionieren (OQ). Ohne eine ordnungsgemäße Installation (IQ) können Sie der Geräteleistung nicht vertrauen. Die Materialqualifizierung (MQ) geht mit der Validierung der Ausrüstung einher, um sicherzustellen, dass alles als vollständiges Verpackungssystem zusammenarbeitet. Wenn dieser systematische Ansatz ordnungsgemäß durchgeführt wird, werden die Validierungsnachweise Schritt für Schritt erstellt, wobei jede Säule die anderen stärkt. Wenn Sie Schritte überspringen, verursachen Sie teure Probleme:
- Wenn Sie den IQ überspringen, wissen Sie nicht, ob Geräteprobleme Ihre späteren Testergebnisse durcheinander bringen.
- Ein schwacher OQ sorgt für Prozessparameter, die beim Testen gut aussehen, aber unter Produktionsstress auseinanderfallen.
- Durch die Qualifizierung von Materialien durch Abkürzungen kommen Teams am Ende zu Verpackungen, die beim Versand versagen oder im Laufe der Zeit an Sterilität verlieren.
Stellen Sie sich die Validierung wie den Bau eines Hauses vor — Sie würden nicht mit dem Dach beginnen, bevor Sie ein solides Fundament gelegt haben. Die vier Säulen funktionieren auf die gleiche Weise, wobei jede Phase das unterstützt, was als nächstes kommt. Wenn Sie die Vorarbeiten überspringen, verbringen Sie viel mehr Zeit und Geld damit, Probleme zu lösen, die mit einem systematischen Ansatz von Anfang an hätten vermieden werden können.
Installationsqualifizierung: Überprüfung und Kalibrierung von Geräten
Ohne eine solide Basisdokumentation ist es unmöglich, Geräteprobleme und Prozessfehler in späteren Validierungsphasen voneinander zu trennen. Die Installationsqualifizierung (IQ) verhindert dies, indem sie vor Beginn der Leistungstests genau festlegt, womit Sie beginnen.
Das zentrale Ergebnis von IQ ist die Dokumentation der Basisparameter, die den Ausgangspunkt Ihrer Ausrüstung erfasst. Sie werden aufnehmen:
- Betriebsbereiche für kritische Parameter wie Temperatur, Druck, Geschwindigkeit und Verweilzeit
- Standardeinstellungen und Steuerungssystemkonfigurationen
- Umgebungsbedingungen im Verpackungsbereich
- Leistungsmerkmale der Ausrüstung unter Leerlaufbedingungen
- Präventive Wartungsanforderungen und Zeitpläne, die eine gleichbleibende Leistung der Geräte gewährleisten
Die Überprüfung und Kalibrierung der Ausrüstung bilden die Grundlage für diese Basislinien. Sie müssen sicherstellen, dass alle Komponenten den Einkaufsspezifikationen entsprechen, dass die Versorgungseinrichtungen die Anforderungen erfüllen und dass die Sicherheitssysteme ordnungsgemäß funktionieren. Bei Dichtungsgeräten dokumentieren Sie die Temperatur- und Druckeigenschaften sowie die technischen Daten der Heizelemente. Steuerungssysteme werden überprüft, um sicherzustellen, dass sie die erforderlichen Parameter einhalten können.
Die Kalibrierung erweitert die Grundlage des IQ. Geräte, die den Verpackungsprozess überwachen oder steuern — Temperaturregler, Manometer, Zeitmesssysteme und Prüfgeräte — müssen über Zertifikate verfügen, die auf nationale Normen rückführbar sind. Diese Aufzeichnungen belegen die Leistung der Geräte und belegen die Prozesskontrolle während der Validierung und der routinemäßigen Produktion.
Dieser Datensatz wird auch zum Bezugspunkt für spätere Etappen. Wenn sich die Dichtheit während der Leistungsqualifizierung verändert, können die Ergebnisse auf Geräteunterschiede zurückgeführt werden, die im IQ dokumentiert sind. Ohne diese umfangreiche Dokumentation wird es schwierig, zwischen Geräteproblemen und Prozessausfällen zu unterscheiden, was zu kostspieligen Untersuchungen führt, die eine gründliche Vorarbeit verhindert hätte.
Betriebliche Qualifizierung: Prüfung in allen Betriebsbereichen
Ihre Ausrüstung läuft unter idealen Bedingungen möglicherweise einwandfrei, liefert aber inkonsistente Ergebnisse, wenn die Produktion chaotisch wird. Die Betriebsqualifizierung (OQ) deckt diese Problembereiche auf, bevor sie zu Problemen im nachgelagerten Bereich führen. Indem die Teams die Parameter an ihren Grenzen testen, können sie erkennen, wo der Prozess stabil ist und wo die Leistung nachlässt.
Ihr Ziel ist es, Prozessparameter festzulegen, die die Produktionsteams täglich verwenden werden — die Betriebsgrenzen mit normalen Sollwerten plus akzeptablen Variationsbereichen. Sie finden Bereiche, in denen die Ausrüstung konstant zuverlässige Ergebnisse liefert, und zwar mit optimalen Einstellungen, die die beste Leistung bieten, und Sicherheitsmargen, die Probleme verhindern. Um die Funktionalität der Geräte zu testen, muss ein Parameter systematisch variiert und andere Parameter konstant gehalten werden. Anschließend wird die Qualität der Verpackung überwacht. Das Testen an den Rändern hilft dabei zu erkennen, wo die Leistung nachlässt. Dies wird zu Ihrer Sicherheitsmarge für die routinemäßige Produktion.
OQ-Aufzeichnungen müssen über die Aufzählung der Funktionen der Geräte hinausgehen. Sie sollten auch die Bedingungen erfassen, unter denen die Ergebnisse konsistent bleiben. So kann beispielsweise eine hohe Luftfeuchtigkeit die Abdichtung beeinträchtigen, Temperaturschwankungen können zu Abweichungen bei kritischen Parametern führen, und in der Nähe befindliche Maschinen können zu Vibrationen führen, die die Präzision beeinträchtigen.
Bei diesem Test werden Parameterkombinationen erkannt, die zu Problemen in der Produktion führen würden, bevor sie tatsächlich auftauchen. Ihre Ausrüstung läuft möglicherweise mit bestimmten Einstellungen, liefert aber inkonsistente Ergebnisse, die nachgelagerte Prozesse Kopfzerbrechen bereiten. OQ identifiziert diese Problembereiche, sodass die Produktionsteams sie meiden können. Die hier festgelegten Bereiche bilden die Grundlage für Bedienerschulungen und Prozessalarme, die für einen reibungslosen Produktionsablauf sorgen.
Leistungsqualifizierung: Nachweis einer gleichbleibenden Verpackungsleistung
Die Leistungsqualifizierung (PQ) zeigt, dass Ihr vollständiger Verpackungsprozess konsistent Verpackungen produziert, die alle Akzeptanzkriterien unter realen Produktionsbedingungen erfüllen. Hier geht die Validierung vom Nachweis der Leistungsfähigkeit zum Nachweis der Zuverlässigkeit über.
Die Realität ist, dass Ihr Verpackungsprozess funktionieren muss, wenn nicht alles perfekt ist. Die Geräteeinstellungen weichen leicht ab, die Bediener wechseln zwischen den Schichten und die Qualität der Materialpartien variiert. Die Umgebungsbedingungen schwanken im Laufe des Tages. PQ-Tests zeigen, dass Ihr Prozess trotz dieser normalen Schwankungen immer noch akzeptable Verpackungen produziert. Tests im schlimmsten Fall gehen noch einen Schritt weiter, indem Sie Ihren validierten Prozess an realistische Grenzen bringen. Stresstests sollten die Realität der Produktion widerspiegeln und nicht nur ideale Bedingungen. Das kann bedeuten, mit maximaler Liniengeschwindigkeit zu fahren, während die Bediener auf die Einhaltung von Terminen drängen, unter reduzierten Umweltkontrollen zu arbeiten, wenn die HLK-Systeme nicht gut funktionieren, oder die Qualität am Ende einer Schicht zu testen, wenn Ermüdung die Ergebnisse beeinflussen kann.
Die Durchführung von Validierungstests unter tatsächlichen Produktionsbedingungen bedeutet, dieselben Materialien, Geräteeinstellungen und Umgebungsbedingungen zu verwenden, mit denen Fertigungsteams täglich konfrontiert sind. Die Erfassung natürlicher Prozessschwankungen im Laufe der Zeit, der Bediener, der Materialchargen und des Gerätezustands ist wichtiger als Tests unter künstlich kontrollierten Bedingungen, die nicht der Realität entsprechen. Die statistische Planung legt fest, wie viele Tests erforderlich sind, um eine gleichbleibende Leistung nachzuweisen. Ein gut durchdachter Stichprobenplan deckt mehrere Durchläufe und Bediener ab und stellt sicher, dass die Ergebnisse die tatsächliche Prozessfähigkeit und nicht den Zufall widerspiegeln. Diese Nachweise dienen dann der Prozessüberwachung und stellen die validierte Leistung während der täglichen Fertigung sicher.
Material- und Designqualifizierung: Gewährleistung von Kompatibilität und Schutz
Die Materialqualifizierung (MQ) zeigt, dass die Verpackungsmaterialien von Geräten während des gesamten Gerätelebenszyklus wie vorgesehen funktionieren können. Bei endsterilisierten Medizinprodukten bedeutet dies, dass eine Primärverpackung ausgewählt werden muss, die der gewählten Sterilisationsmethode standhält und die Sterilität während des Vertriebs und der Lagerung weiterhin gewährleistet. Die Validierung des Verpackungssystems bestätigt dann, dass Materialien, Design und Versiegelung während der gesamten vorgesehenen Lebensdauer des Geräts als sterile Barriere wirken.
Bei der Auswahl der Materialien müssen die Teams drei Faktoren berücksichtigen:
- Die Sterilisationsmethode, die verwendet wird
- Die Speicherumgebung, der das Paket standhalten muss
- Die Benutzeranforderungen für sicheres Öffnen und Gerätezugriff
Jede Sterilisationsmethode ist mit einzigartigen Herausforderungen verbunden. Für die Ethylenoxid-Sterilisation sind in der Regel Materialien erforderlich, die chemischem Abbau widerstehen und gleichzeitig das Eindringen von Gas und die Evakuierung ermöglichen, um eine effektive Sterilisation und Belüftung zu gewährleisten. Durch die Gammasterilisation werden Verpackungen Strahlung ausgesetzt, die ihre Festigkeit und Flexibilität beeinträchtigen kann. Bei der Dampfsterilisation werden hohe Temperaturen mit Feuchtigkeit kombiniert, wodurch die Barriereeigenschaften beeinträchtigt werden können. Wenn diese Risiken nicht frühzeitig behoben werden, kann die Sterilbarriere im realen Gebrauch versagen, wodurch sowohl die Einhaltung der Vorschriften als auch die Patientensicherheit gefährdet werden.
Bei Materialverträglichkeitstests wird bewertet, wie Ihre Verpackung während der gesamten vorgesehenen Haltbarkeit mit den Gerätekomponenten interagiert. Dabei muss jedoch auch die praktische Verwendbarkeit berücksichtigt werden. Die Materialien müssen außerdem ihre mikrobiellen Barriereeigenschaften beibehalten, um sicherzustellen, dass sterile Barrieren während der gesamten vorgesehenen Haltbarkeitsdauer Kontaminanten blockieren. Gleichzeitig muss die Schälfestigkeit stark genug sein, um die Unversehrtheit der Versiegelung während der Verteilung aufrechtzuerhalten, gleichzeitig aber auch so sanft sein, dass das medizinische Fachpersonal das Produkt unter aseptischen Bedingungen öffnen kann, ohne dass es reißt. Sie führen Studien zur beschleunigten Hautalterung durch, in denen jahrelange Lagerung in Wochen simuliert wird. Kombiniert mit der Alterung unter tatsächlichen Lagerbedingungen in Echtzeit, um Ihre Prognosen zu bestätigen.
Protokolle zur Materialprüfung:
Erste Testprotokolle helfen dabei, die Ausgangsleistung des Materials zu ermitteln, bevor vollständige Validierungsstudien beginnen. Konzentrieren Sie sich auf die Entwicklung von Testprotokollen, die Materialien so beanspruchen, wie sie im realen Einsatz tatsächlich beansprucht werden, anstatt willkürliche Testspezifikationen zu erfüllen, die sich nicht auf die tatsächlichen Leistungsanforderungen beziehen. Die Öffnungseigenschaften sollten einen sauberen, kontrollierten Zugang ermöglichen, ohne die Sterilität zu beeinträchtigen oder Partikel in das sterile Feld zu streuen. Das bedeutet, dass Ihre Tests die tatsächlichen klinischen Einsatzbedingungen simulieren müssen und nicht ideale Laborumgebungen.
Vertriebs- und Transittests: Simulation der realen Schifffahrt
Die Verpackung muss den gesamten Transport von der Produktionshalle bis zum Patientenbett überstehen. Bei den Verteilungstests werden die Belastungen beim Versand und der Handhabung nachempfunden, sodass die Teams sicherstellen können, dass die sterilen Barrieren in der gesamten Lieferkette intakt und funktionsfähig sind.
ASTM D 4169 und ISTA-Protokolle skizzieren Sie, wie Sie Vertriebsumgebungen simulieren können, aber Sie müssen sie an Ihre tatsächlichen Versandbedingungen anpassen. Anstatt einzelne Kontrollen durchzuführen, sollten die Tests den gesamten Transportweg berücksichtigen: Ein Paket kann fallen gelassen, durch Transportvibrationen erschüttert oder unter der Ladung des Lagers zerquetscht werden. Unterwegs stellen Orientierungswechsel, wiederholte Handhabung und wechselnde Klimabedingungen die Festigkeit der Sterilbarriere auf die Probe. Wenn man diese Belastungen nacheinander betrachtet, erhält man ein genaueres Bild davon, wie sich Verpackungen in der realen Verteilung verhalten.
Pakete werden nach dem Verteilungstest mehreren Prüfungen unterzogen. Bei der Sichtprüfung wird nach Oberflächenschäden gesucht, während Festigkeitstests bestätigen, dass die Verschlüsse intakt bleiben. Bei empfindlicheren Methoden wie Blasentests werden Mikrolecks in sterilen Barrieren aufgedeckt, die mit dem Auge nicht sichtbar sind. Der letzte Schritt sind Integritätstests, bei denen nachgewiesen wird, dass die mikrobielle Barriere auch nach dem simulierten Versand noch funktioniert. Diese Ergebnisse sind nur wichtig, wenn die Tests die tatsächlichen Versandbedingungen widerspiegeln. Pakete, die generische Protokolle erfüllen, aber im Feld versagen, führen zu Rückrufen und schädigen gleichzeitig den Ruf des Unternehmens. Am wichtigsten ist, dass sie die Patienten einem Risiko aussetzen.

Studien zur beschleunigten Alterung in Echtzeit: Nachweis von Haltbarkeitsangaben
Haltbarkeitsangaben stehen und fallen nur dann, wenn nachgewiesen wird, dass Verpackungen ihre Sterilbarriere so lange aufrechterhalten, wie die Geräte auf dem Markt bleiben. Die beschleunigte Alterung gibt Herstellern einen frühen Überblick über die Leistung von Verpackungen, indem sie erhöhte Temperatur und Luftfeuchtigkeit verwenden, um jahrelange Lagerung in nur wenigen Wochen zu simulieren. ASTM F 1980 (aktuelle Version) beschreibt den Ansatz, aber die eigentliche Schwierigkeit besteht darin, Bedingungen so zu entwerfen, dass sie die tatsächlichen Umgebungen widerspiegeln, ohne Ausfallarten einzuführen, die in der Praxis niemals vorkommen würden. Wenn diese Studien gut konzipiert sind, liefern sie vorläufige Daten, die die ursprünglichen Angaben zur Haltbarkeit stützen und dazu beitragen, dass Produkte weiterentwickelt werden, solange längerfristige Studien noch im Gange sind.
Altern in Echtzeit rundet das Bild ab. Verpackungen aus den letzten Produktionsläufen werden unter realen Bedingungen gelagert und über Monate oder Jahre auf ihre Leistung hin überwacht. Diese Daten belegen, dass Verpackungen den täglichen Anforderungen der Lagerung und des Vertriebs standhalten, und liefern die von den Aufsichtsbehörden erwarteten bestätigenden Daten. Teams, die frühzeitig mit Echtzeitstudien beginnen, vermeiden Datenlücken, die spätere Genehmigungen oder Verlängerungen verzögern könnten.
Die zuverlässigsten Haltbarkeitsbestimmungen ergeben sich aus der Kombination beschleunigter Prognosen mit Bestätigung in Echtzeit. Statistische Analysen zeigen, wann die Verpackungseigenschaften unter Ihre Zielvorgaben fallen, und geben Ihnen Konfidenzintervalle für Angaben zur Haltbarkeit. Eine schwache Planung kann jedoch den gesamten Aufwand zunichte machen — das Testen von Prototypen anstelle von Produktionsläufen, die Auswahl von Lagerbedingungen, die nicht der Realität entsprechen, oder die Unterschätzung der Stichprobengrößen untergraben die Glaubwürdigkeit.
Mit dem richtigen Ansatz belegen Alterungsstudien, dass Verpackungen die Patienten über die gesamte vorgesehene Lebensdauer hinweg schützen und gleichzeitig Herstellern und Aufsichtsbehörden die Gewissheit geben, dass die Angaben im Laufe der Zeit Bestand haben werden. Diese Nachweise gehen über die regulatorischen Anforderungen hinaus und belegen die Leistung in der Praxis, die für die Patientensicherheit von Bedeutung ist.
Bewertung der Benutzerfreundlichkeit und menschliche Faktoren: Validierung der Benutzerinteraktion
EIN Verpackungssystem kann alle technischen Standards erfüllen und trotzdem versagen, wenn es in den Händen eines Arztes nicht sicher geöffnet werden kann. Die Überprüfung der Benutzerfreundlichkeit stellt sicher, dass sterile Barrieresysteme nicht nur im Labor funktionieren, sondern auch in der Praxis, wenn Geräte für die Patientenversorgung geöffnet werden. Sowohl die FDA als auch ISO 11607 verknüpfen die Benutzerfreundlichkeit direkt mit dem Risikomanagement und erkennen an, dass Verpackungen, bei denen die aseptische Technik beeinträchtigt wird, praktisch ein System sind, das versagt.
Ein zentraler Schwerpunkt ist aseptischer Präsentationstest - Nachweis, dass die Sterilbarriere in der Praxis sicher geöffnet werden kann. Verschlusssysteme und Verpackungsverschlüsse müssen einem vorzeitigen Ausfall standhalten und sich dennoch unter klinischen Bedingungen problemlos öffnen lassen. Die Integrität der Versiegelung ist unerlässlich. Verpackungen müssen sich sauber abziehen lassen, ohne zu reißen, und das Design muss verhindern, dass Partikel oder Schmutz in das sterile Feld gelangen. Ein häufiges Problem ist die überdimensionierte Versiegelung. Sie ist stark genug, um die Testziele zu erfüllen, aber so schwer zu öffnen, dass eine aseptische Technik nicht praktikabel ist. Simulationstests helfen dabei, diese Probleme zu lösen, bevor Produkte auf den Markt kommen.
In der Praxis öffnen Krankenschwestern oder Operationstechniker häufig die Verpackungen unter sterilen Bedingungen, während die Beobachter etwaige technische Störungen feststellen. Der Handschuhtyp ist wichtig: Nitril und Latex bieten unterschiedliche Griffigkeit und Haptik. Andere reale Faktoren — wie die Beleuchtung im OP, der Zeitdruck bei Eingriffen oder die Art und Weise, wie gut die Verpackung selbst zusammenarbeitet — wirken sich ebenfalls auf die Fähigkeit aus, die Sterilität aufrechtzuerhalten. Sogar Details wie Abziehlaschen können den Unterschied ausmachen: Mit bloßen Händen leicht zu handhaben, mit Handschuhen fast unmöglich. Eine zu starke Versiegelung birgt das gleiche Risiko und zwingt zu Behelfslösungen, die den gesamten Zweck steriler Verpackungen untergraben. Eine Versiegelung, die nicht unter aseptischen Bedingungen geöffnet werden kann, frustriert nicht nur die Anwender, sondern beeinträchtigt auch direkt die Sterilität am Behandlungsort.
Ebenso wichtig ist Verständnis wie Menschen in der Praxis mit dem Paket umgehen. In Studien zur Benutzerfreundlichkeit wird beobachtet, wie Gesundheitsdienstleister Geräte unter realistischen Bedingungen öffnen und dabei Momente des Zögerns sowie technische Abweichungen oder ein potenzielles Kontaminationsrisiko auftauchen. Diese Beobachtungen zeigen Probleme auf, die bei Materialtests übersehen werden — Versiegelungen, die zu viel Kraft erfordern, oder Verpackungen, die unvorhersehbar reißen, wenn Benutzer schnell darauf zugreifen müssen. Unklare Hinweise beim Öffnen sorgen für Verwirrung, was die sterile Technik bei den eigentlichen Eingriffen beeinträchtigt.
Schließlich muss die Validierung Folgendes berücksichtigen Anforderungen an die Ausbildung. Wenn die sichere Verwendung von komplexen Anweisungen abhängt, steigt das Fehlerrisiko. Eine frühzeitige Bewertung des Schulungsbedarfs hilft zu bestätigen, dass die Verpackung für die vorgesehenen Benutzer intuitiv zu bedienen ist. Alle Anweisungen, die für eine sichere Handhabung unerlässlich sind, sollten klar dokumentiert werden. Teams, die diesen Schritt von Anfang an unternehmen, sind besser positioniert, um kostspielige Umgestaltungen oder unerwartete Beschwerden nach der Markteinführung zu vermeiden.
Wenn die Benutzerfreundlichkeit von Anfang an in die Validierung integriert wird, erhalten Hersteller mehr als nur Konformitätsnachweise. Sie liefern Verpackungen, die sowohl für Patienten als auch für Ärzte geeignet sind, und stellen gleichzeitig sicher, dass die Aufsichtsbehörden dort funktionieren, wo es am wichtigsten ist.
Qualifizierung der Prozessleistung: Validierung der Fertigungskonsistenz
Selbst die sorgfältig entworfene Verpackung versagt, wenn der Herstellungsprozess nicht jedes Mal das gleiche Ergebnis liefert. Die Prozessleistungsqualifizierung (PPQ) beweist, dass Verpackungsläufe nicht nur im Labor oder bei Validierungsversuchen erfolgreich sind, sondern auch im vollen Produktionsmaßstab konsistent bleiben. Dies ist der Punkt, an dem sowohl Aufsichtsbehörden als auch interne Qualitätsteams den Nachweis benötigen, dass Verpackungssysteme der realen Produktion standhalten.
Ein starker PPQ untersucht den gesamten Produktionsablauf, einschließlich der Montageprozesse. Dabei wird untersucht, wie Materialien gelagert werden, wie Bediener mit Komponenten umgehen und ob Umweltkontrollen für stabile Bedingungen sorgen. Jede Schwachstelle in diesen Bereichen kann die Leistung der Verpackung beeinträchtigen, selbst wenn die Geräteeinstellungen eingegeben werden.
Zuverlässigkeit entsteht durch Wiederholbarkeit. Aus diesem Grund erfordert PPQ mehrere Produktionsläufe, um nachzuweisen, dass der Prozess über verschiedene Chargen hinweg konsistente Ergebnisse liefert. Teams validieren in der Regel drei aufeinanderfolgende Chargen, um nachzuweisen, dass die Leistung in den verschiedenen Produktionsläufen konstant ist. Die Anforderungen können jedoch je nach Produktrisiko und behördlichen Vorgaben variieren. Durch kontinuierliche Überwachung wird diese Sicherheit gewährleistet und Abweichungen werden erkannt, bevor daraus Qualitätsprobleme werden.
Der Nutzen geht weit über die Einhaltung von Vorschriften hinaus. Ein gut ausgeführtes PPQ bietet Vorteile, die weit in den täglichen Betrieb hineinreichen. Es macht die Produktion vorhersehbarer und beschleunigt gleichzeitig Release-Entscheidungen. Vor allem aber schafft es bei den Aufsichtsbehörden das Vertrauen, dass das System unter Kontrolle ist.
Revalidierungsanforderungen und Änderungsmanagement
Veränderungen sind in der Fertigung unvermeidlich, und jede Änderung wirft die Frage auf, ob Ihr Verpackungssystem noch seinen Zweck erfüllt. Eine erneute Validierung belegt, dass dies der Fall ist, und verhindert, dass kleine Anpassungen zu großen Risiken führen. Die Aufsichtsbehörden betrachten unkontrollierte Veränderungen als Alarmsignal, und das sollten auch die Hersteller tun. Zu den häufigsten Auslösern gehören:
- Geräte-Updates, z. B. neue Dichtungs- oder Inspektionssysteme
- Änderungen bei der Sterilisation, sei es durch eine Änderung der Methode oder durch Parameteranpassungen
- Materialmodifikationen, vom Lieferantenwechsel bis zur Auswahl verschiedener Substrate
- Prozessänderungen, auch geringfügige, die die Betriebsbedingungen verändern
Der Umfang der Revalidierung hängt vom Risiko und den dokumentierten Validierungsanforderungen ab. Jede Änderung eines Prozesses wirft die Frage der Sterilität auf. Teams stellen möglicherweise fest, dass eine kleine Anpassung die Integrität der Siegel beeinträchtigt. In anderen Fällen muss die Materialverträglichkeit neu bewertet werden. Manchmal geht es um die Frage, ob die mikrobiellen Barriereeigenschaften noch bestehen. Was auch immer der Auslöser sein mag, Folgetests liefern den Nachweis, dass die Verpackung weiterhin wie vorgesehen schützt. Regelmäßige Überprüfungen werden ebenfalls erwartet, da sich die Ausrüstung mit der Zeit abnutzt. Lieferantenwechsel oder allmähliche Prozessabweichungen können die gleichen Auswirkungen haben, und jedes muss überwacht werden.
Eine gute Dokumentation fasst all dies zu Beweisen zusammen, denen die Aufsichtsbehörden vertrauen können. Sie sollte die Gründe für jeden Test erläutern und nicht nur festhalten, dass der Test stattgefunden hat. Klare Aufzeichnungen bieten mehr als nur Unterlagen. Sie geben den Aufsichtsbehörden Vertrauen, helfen Teams dabei, Änderungen proaktiv zu bewältigen, und vor allem zeigen sie, dass Verpackungen Patienten auch nach dem ersten Validierungsbericht schützen.
Gemeinsame Herausforderungen und effektive Lösungen
Die Validierung kann aus bekannten Gründen ins Stocken geraten. Stichprobengrößen werden oft unterschätzt, Worst-Case-Tests werden übersprungen und die Dokumentation verrutscht, bis sie nicht mehr dem entspricht, was gemacht wurde. Die Teams verbringen außerdem Stunden damit, Dokumente manuell zu vergleichen und zu hinterfragen, ob sie jede wichtige Änderung erkannt haben. Jeder dieser Fehltritte mag sich im Moment klein anfühlen, aber zusammen verursachen sie kostspielige Nacharbeiten und führen zu behördlichen Feststellungen, die der Glaubwürdigkeit auch noch lange nach Ende des Audits schaden. Der Engpass bei der manuellen Überprüfung ist besonders problematisch, weil er erfahrene Qualitätsexperten in langwierigen Vergleichen überfordert, anstatt ihr Fachwissen für die Risikobewertung und strategische Entscheidungsfindung einzusetzen.
Der stärkere Ansatz besteht darin, die Validierung als strategische Aktivität aufzubauen. Klare Akzeptanzkriterien, die von Anfang an festgelegt werden, tragen dazu bei, Unstimmigkeiten in der Spätphase zu vermeiden, insbesondere wenn sich die Teams für Qualität, Technik und Regulierung früh im Prozess abstimmen. Wenn die Validierung während der gesamten Entwicklung stattfindet und nicht als letzter Schritt behandelt wird, lassen sich Lücken leichter erkennen und behördliche Prüfungen laufen reibungsloser ab.
Effizient zu arbeiten bedeutet, sich dort einzusetzen, wo es am meisten zählt, und nicht nur Abstriche zu machen. Die risikobasierte Stichprobe konzentriert sich auf Ressourcen, bei denen ein Ausfall am wichtigsten wäre. Testpläne können so konzipiert werden, dass sie mehr als eine Anforderung gleichzeitig erfüllen, wodurch Redundanzen vermieden werden, ohne die Strenge zu verringern. Auf diese Weise sparen Best Practices Zeit und stärken gleichzeitig die Evidenzbasis. So wird sichergestellt, dass die Verpackung das Vertrauen der Aufsichtsbehörden weckt und die Patienten, die sie verwenden, auch weiterhin schützt.
Dokumentation und behördliche Einreichung
Für die Aufsichtsbehörden ist es nicht passiert, wenn es nicht dokumentiert ist. Hersteller sehen sich mit der gleichen Realität konfrontiert: Unvollständige Aufzeichnungen können Genehmigungen verzögern und gleichzeitig Inspektionsergebnisse auslösen, die das Vertrauen in das Qualitätssystem schwächen.
Eine solide Dokumentation schließt diese Lücken. Eine vollständige Aufzeichnung zeigt, wie die Tests geplant und durchgeführt wurden, und verknüpft die Ergebnisse mit den Akzeptanzkriterien. Diese Dokumentation belegt die Einhaltung gesetzlicher Vorschriften. Validierungszertifikate und technische Unterlagen formalisieren diese Nachweise und bestätigen, dass Verpackungssysteme sowohl die Anforderungen von ISO 11607 als auch die der FDA erfüllen. Eine effektive Dokumentation ist mehr als ein Sammelband mit Berichten. Sie vereint Protokolle, Daten und Risikobeurteilungen zu einer kontrollierten, inspektionsbereiten Compliance-Dokumentation. Wenn die Aufsichtsbehörden danach fragen, müssen die Hersteller nicht nur nachweisen, was getestet wurde, sondern auch warum, wie und mit welchem Ergebnis.
Eine gründliche Dokumentation erfüllt nicht nur die Anforderungen der Aufsichtsbehörden, sondern sorgt auch dafür, dass Geräte ohne kostspielige Unterbrechungen auf den Markt gebracht werden können. Wenn die Aufzeichnungen vollständig und inspektionsbereit sind, können Hersteller ihren Prozess mit Zuversicht verteidigen, und die Patienten erhalten eine Verpackung, die sie während des gesamten Lebenszyklus des Geräts schützt.
Sicherstellung des Erfolgs des Verpackungssystems
Die Validierung von Verpackungen beweist, dass sterile Barrieresysteme dem Transport von der Fabrikhalle zum Behandlungsort standhalten können. Die Validierung basiert auf einer sorgfältigen Planung und wird durch solide Tests und Unterlagen untermauert. Sie gibt den Herstellern die Gewissheit, dass ihre Verpackungen vor Ort funktionieren und die Anforderungen der Aufsichtsbehörden erfüllen. Wenn die Validierung als Lebenszyklusaufgabe und nicht als einmalige Aufgabe betrachtet wird, werden kostspielige Fehler vermieden und das Vertrauen der Qualitätsteams und der Aufsichtsbehörden gleichermaßen gestärkt.
Die Landschaft entwickelt sich. Digitale Validierungsplattformen und engere Verbindungen zum Risikomanagement führen zu einer Neugestaltung der Best Practices, während die Aufsichtsbehörden eine stärkere globale Abstimmung anstreben. Was sich nicht ändern wird, ist das Ziel: sicherzustellen, dass die Verpackung steril bleibt, sodass sich Ärzte auf jedes Gerät verlassen können und die Patienten den Schutz erhalten, den sie verdienen. Mit Blick auf die Zukunft wird eine engere Verzahnung der Verpackungsvalidierung mit der Sterilisationsvalidierung die Praxis in der Branche prägen und sicherstellen, dass Angaben zur Sterilität lückenlos unterstützt werden.
Verify von GlobalVision beschleunigt die Validierung von Verpackungen, indem die Prüfung von Dokumenten und Kunstwerken automatisiert wird, sodass die Teams sich auf die Einhaltung der Vorschriften verlassen können, ohne dass dies zu Verzögerungen führt.
