
Selbst eine einzige umgeformte Ziffer auf einem Arzneimitteletikett kann Patienten einem Risiko aussetzen und zu kostspieligen Rückrufen führen. Die Unternehmen, die bereit für Inspektionen sind, kennen nicht nur die Anforderungen, sondern wissen auch, wo Fehler am häufigsten auftreten.
Einführung in die Arzneimittelkennzeichnung
Die pharmazeutische Kennzeichnung umfasst jedes Element, das auf der Arzneimittelverpackung erscheint. Dies umfasst nicht nur die Wörter, sondern auch grafische Komponenten wie Logos, Farbbänder, Barcodes, Brailleschrift und behördliche Symbole in allen Bereichen, vom Text der Durchstechflasche über Verschreibungsinformationen bis hin zu Werbematerial und Packungsbeilagen. Die Definition der FDA gemäß der Bundesgesetz über Lebensmittel, Arzneimittel und Kosmetik wirft ein breites Netz aus - wenn es Ihrem Medikament beiliegt, ist es die Kennzeichnung. Aber verschiedene Märkte haben unterschiedliche Regulierungsbehörden. In den USA wird es vom Zentrum für Arzneimittelbewertung und -forschung der FDA verwaltet. Europa weist die Aufgabe der EMA zu, während Japan auf die PMDA angewiesen ist. Der Internationale Harmonisierungsrat (ICH) hat die Unterschiede durch Harmonisierung verringert, aber die eigentliche inhaltliche Harmonisierung ist noch nicht abgeschlossen. Es bestehen weiterhin regionale Unterschiede, nicht nur in Bezug auf Sprache und Übersetzung, sondern auch in Bezug auf die Datenanforderungen, die Reihenfolge der Inhalte und die lokalen Nutzen-Risiko-Prioritäten. Das bedeutet, dass ein Etikett, das von der FDA geprüft wird, möglicherweise noch überarbeitet werden muss, bevor es von den europäischen Aufsichtsbehörden akzeptiert wird.
Die Arzneimittelkennzeichnung umfasst auch rezeptfreie Medikamente (OTC), die über ein eigenes standardisiertes Etikettenformat mit „Drug Facts“ verfügen. Im Gegensatz zu verschreibungspflichtigen Etiketten, die für Gesundheitsdienstleister geschrieben werden, muss die OTC-Etikettierung für Verbraucher selbsterklärend sein. Die Typografieregeln für die OTC-Etikettierung sind ungewöhnlich streng. Die Drug Facts-Verordnung der FDA schreibt vor, dass der Text in mindestens 6-Punkt-Schrift mit fett gedruckten Überschriften und einem Layout versehen sein muss, das auf Armlänge lesbar bleibt. Innerhalb dieses Formats legt die FDA auch fest, welche Elemente in dieser Reihenfolge erscheinen müssen:
- Wirkstoff (e): Name und Stärke pro Dosierungseinheit
- Zweck: die Arzneimittelkategorie oder die beabsichtigte Wirkung
- Verwendungen: zugelassene Indikationen
- Warnungen: einschließlich Hinweisen „Nicht verwenden“, Vorsichtsmaßnahmen für Schwangerschaft/Stillzeit und wann ein Arzt konsultiert werden muss
- Anfahrt: Dosierungsanweisungen für den sicheren Gebrauch
- Inaktive Inhaltsstoffe: zur Sensibilisierung für Allergien gelistet
Unklare Dosierungsanweisungen und schlecht platzierte Gegenanzeigen führen dazu, dass Verbraucher nicht alle Informationen haben, die sie für eine sichere Anwendung der Produkte benötigen. Weil das Risiko von Missverständnissen so hoch ist, verschärft sich die regulatorische Kontrolle ständig.
Ressourcen zur Kennzeichnung verschreibungspflichtiger Medikamente durch die FDA
Die FDA betreibt mehrere Datenbanken für den Zugriff auf offizielle Kennzeichnungsinformationen. Medikamente @FDA ist die umfassendste und enthält Zulassungsinformationen, Verschreibungsdetails und Kennzeichnungshistorie für Tausende zugelassener Medikamente. Möchten Sie sehen, wie sich ein Etikett entwickelt hat? Hier suchen Sie.
Tägliches Med, das von der National Library of Medicine zusammen mit der FDA betrieben wird, veröffentlicht Standardproduktkennzeichnungen in strukturiertem Format. Gesundheitsdienstleister verwenden es, wenn sie die Verschreibungsinformationen am Behandlungsort überprüfen. Wenn Sie auf beiden Portalen navigieren, beginnen Sie mit dem Abschnitt „Highlights“, in dem wichtige Verschreibungsinformationen zusammengefasst sind, bevor Sie die ausführlichen Abschnitte zur klinischen Pharmakologie und zu den Aufbewahrungsanforderungen besuchen.
Bei behördlichen Inspektionen vergleichen Auditoren die Produktionsetiketten mit offiziellen Unterlagen. Die von der FDA zugelassene Version entspricht dem, was die Behörde geprüft und freigegeben hat, während die aktuelle Kennzeichnung die neuesten Aktualisierungen widerspiegelt. Qualitätsteams sollten die von der FDA zugelassene Version als Compliance-Benchmark verwenden, nicht das, was gerade auf DailyMed steht. Diese Unterscheidung ist wichtig, wenn Inspektoren mit Ihren Einreichungsdokumenten in der Hand erscheinen.
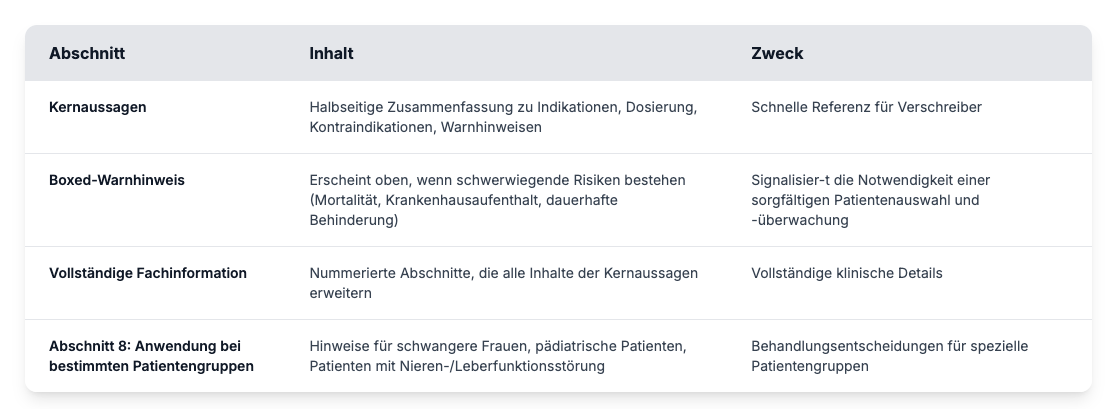
Bestandteile der Verschreibungsinformationen
Professionelle Verschreibungsinformationen folgen dem Format der Physician Labeling Rule, das den Inhalt in zwei Hauptabschnitte unterteilt: Highlights und vollständige Verschreibungsinformationen.

Die Kennzeichnung von pädiatrischen Patienten erfordert besondere Überlegungen, die über die Angaben in Abschnitt 8 der Verschreibungsinformation hinausgehen. Gemäß dem Best Pharmaceuticals for Children Act (BPCA) und dem Pediatric Research Equity Act (PREA) müssen Hersteller pädiatrische klinische Daten einreichen, anstatt sich ausschließlich auf Studien an Erwachsenen zu verlassen. In der Praxis werden bei der Kennzeichnung von Kindern gewichtsabhängige Dosierungstabellen mit klaren Anweisungen zu altersgerechten Formulierungen kombiniert. Außerdem werden Sicherheitswarnungen angezeigt, wenn die Risiken den potenziellen Nutzen überwiegen. Diese Angaben tragen dazu bei, eine unangemessene Anwendung außerhalb der Zulassungsvorschriften zu verhindern, eine der Hauptursachen für Dosierungsfehler bei Kindern, und geben Gesundheitsdienstleistern Informationen an die Hand, die sie direkt in der Praxis anwenden können.
Bei Daten aus klinischen Studien müssen die Verschreibungsinformationen über die Schlagzeilergebnisse hinausgehen. Damit die Kennzeichnung aussagekräftig ist, muss sie eine absolute Risikoreduktion in einer Weise darstellen, die den Merkmalen der Studienpopulation und der Dauer der Nachbeobachtung Rechnung trägt. Ohne diesen Kontext können Anbieter nicht beurteilen, ob die Studienergebnisse für ihre Patienten zutreffen. In Krankenhäusern ziehen Systeme zur klinischen Entscheidungsunterstützung diese Daten ab, um potenzielle Probleme zu erkennen, bevor Medikamente die Patienten erreichen.
Anforderungen an die Kennzeichnung von Patienten
Bei der Kennzeichnung von Patienten werden medizinische Informationen verwendet und für Personen ohne klinische Ausbildung zugänglich gemacht. Die FDA schreibt je nach Arzneimittelklasse und Risikoprofil spezifische Formate vor: Arzneimittelleitfäden, Packungsbeilagen für Patienten und Anwendungshinweise. Arzneimittelleitfäden sind obligatorisch, wenn Informationen zur Vermeidung schwerwiegender Nebenwirkungen für eine sichere Anwendung von entscheidender Bedeutung sind. In diesen einseitigen Dokumenten werden Risiken in einfacher Sprache erläutert, häufig im Frage-Antwort-Format. Die FDA genehmigt vorab den genauen Wortlaut, was bedeutet, dass Hersteller Inhalte ohne behördliche Genehmigung nicht ändern können.
Eine klare Kommunikation bei der Kennzeichnung geht über die einfache Auswahl des Wortschatzes hinaus. Die FDA erwartet klare Sprache — aktive Stimme statt passiver Konstruktionen, kurze Sätze vor komplexen, alltägliche Terminologie vor medizinischem Fachjargon. Wenn technische Begriffe unvermeidlich sind, müssen sofort Definitionen in einfacher Sprache folgen. Untersuchungen zeigen, dass Patienten mit größerer Wahrscheinlichkeit Dosierungsanweisungen befolgen und Warnzeichen erkennen, wenn das Etikett ihre Sprache spricht.
Das Layout spielt eine ebenso wichtige Rolle für das Verständnis der Patienten. Seiten, die sich offen und strukturiert anfühlen, sind einfacher zu navigieren, und starke Überschriften führen die Leser zu den Details, die sie benötigen. Bei älteren Erwachsenen macht ein größerer Typ oft den Unterschied zwischen klaren Anweisungen und vergessenen Dosen aus. Um dies zu verstärken, schreibt die FDA Mindestschriftgrößen und einen hohen Kontrast zwischen Text und Hintergrund vor. Dabei ist man sich bewusst, dass die Kennzeichnung nur funktioniert, wenn die Patienten sie tatsächlich lesen können. Viele Teams verwenden jetzt automatisierte Tools zur Überprüfung der Schriftgröße um die Einhaltung der Vorschriften sicherzustellen, bevor die Verpackung in die Produktion gelangt.
Standards für die Etikettierung von Kartons und Behältern
Die erste Verpackungsschicht ist der Behälter, der das Medikament selbst berührt, und er muss die für eine sichere Verwendung erforderlichen Angaben enthalten. Eine zweite Schicht, beispielsweise Kartons oder Versandkartons, unterstützt die Lieferkette, indem sie zusätzliche Informationen anzeigt.
Die FDA spezifiziert den Inhalt, der für beide erforderlich ist:
Primärbehälter:
- Name des Arzneimittels
- Stärke
- Art der Verabreichung
- Chargennummer
- Verfallsdatum
Sekundärverpackung:
- Vollständiger Produktname
- Aktive Inhaltsstoffe
- Darreichungsform
- Nettomenge
- Bedingungen der Lagerung
- Angaben zum Hersteller
- Nationaler Drogenkodex (NDC)
Platzbeschränkungen für kleine Behälter stellen eine praktische Herausforderung dar. Wenn der Behälter zu klein ist, um alle erforderlichen Elemente aufzunehmen, erlaubt die FDA, dass bestimmte Angaben nur auf dem Karton erscheinen, solange die beiden während der Verteilung zusammen bleiben. Der National Drug Code (NDC) ist in diesem Zusammenhang von entscheidender Bedeutung. Er dient als eindeutiges Identifikationsmerkmal, das die Verpackung mit der FDA-Registrierung verknüpft. Dies reduziert das Risiko von Medikamentenverwechselungen in Apotheken und klinischen Einrichtungen.
Sicherheitsinformationen werden nicht für alle Produkte einheitlich dargestellt. Lagerungsbedingungen wie Kühlung oder Schutz vor Licht werden deutlich hervorgehoben, wenn produktspezifische Hinweise dies erfordern. Für kontrollierte Substanzen gelten eigene Regeln: Die DEA schreibt vor, dass das Symbol in der Liste (I—V) gut sichtbar auf dem Etikett erscheint, und zwar in einer größeren Menge als der Arzneimittelname. Wenn das Symbol durch die Verpackung hindurch sichtbar bleibt, muss es nicht separat auf den Umkartons angebracht werden. Barcodes decken eine weitere Risikoebene ab, indem sie die Rückverfolgbarkeit entlang der gesamten Lieferkette ermöglichen und sowohl die FDA- als auch die DEA-Anforderungen verschärfen.
Die Anforderungen an die Barcode-Etikettierung haben sich parallel zu elektronischen Medikationssystemen weiterentwickelt. Gemäß 21 CFR 201.25 verlangt die FDA, dass viele Etiketten verschreibungspflichtiger Medikamente einen Barcode enthalten, der mindestens den NDC codiert. In der Praxis scannen Apotheken und klinische Systeme diesen Barcode bei der Abgabe oder Verabreichung, um das Produkt zu bestätigen und Abweichungen zu erkennen, die bei manueller Transkription übersehen werden könnten.
[Visuell: Beschrifteter Arzneimittelkarton mit NDC-Platzierung, Barcodespezifikationen, Positionierung der Warnhinweise, Formatierung von Chargen-/Verfallsdatum, Anzeige des Lagerzustands]
Überlegungen zur Kennzeichnung von Generika
Die Kennzeichnung von Generika folgt einem Gleichheitsstandard mit spezifischen Ausnahmen. Die FDA verlangt von Generika, dass sie in Bezug auf die wesentlichen Merkmale der Referenzarzneimittel entsprechen müssen: Indikationen, Dosierung, Verabreichungsweg, Stärke und Wirkstoff.
Der klinische Inhalt eines generischen Etiketts muss mit dem Referenzarzneimittel (RLD) identisch sein. Dazu gehören Warnhinweise und Vorsichtsmaßnahmen sowie die Formulierung von Nebenwirkungen und Arzneimittelwechselwirkungen. Wenn die Marke einen Warnhinweis in einem Kästchen trägt, z. B. einen Hinweis auf kardiovaskuläre Risiken, wiederholt das Generikum den Warnhinweis an derselben Stelle und im gleichen Format. Die Bereiche, in denen Generika Spielraum haben, sind geschäftsbezogen. Ein Hersteller kann seinen eigenen Namen und seine Kontaktinformationen verwenden, den Abschnitt „Wie geliefert“ an die Verpackung anpassen und Patent- oder Exklusivitätserklärungen, die nur für das Markenprodukt gelten, weglassen.
Die FDA betont weiterhin, dass die Kennzeichnung von Generika stets den aktuellen Sicherheitsinformationen entspricht. Gemäß Abschnitt 505 (o) (4) kann die Behörde Aktualisierungen verlangen, wenn neue Risiken festgestellt werden. Dadurch wird sichergestellt, dass sowohl das Referenzarzneimittel als auch die zugelassenen Generika mit denselben Warnhinweisen versehen sind. Während die FDA die Aufsicht verschärft, wurde der Mechanismus für eine schnelle Anpassung an die Sicherheitsvorschriften einer Reform unterzogen, und der Zeitpunkt der Aktualisierungen kann in der Praxis variieren. Ein Teil der Angleichung hinkt trotz regulatorischer Akzente immer noch hinterher.
Anforderungen an die Kennzeichnung biologischer Produkte
Biologische Produkte unterscheiden sich in wichtigen Punkten von herkömmlichen Medikamenten. Traditionelle Medikamente werden durch vorhersehbare chemische Prozesse synthetisiert. Biologika werden in lebenden Systemen (Bakterienzellen, Hefe, Zellkulturen von Säugetieren) hergestellt, wobei die Produkteigenschaften zwischen den Produktionschargen variieren können, was sich auf Sicherheit und Wirksamkeit auswirkt.
Ein Biosimilar ist ein biologisches Produkt, das einem bereits zugelassenen Referenzprodukt sehr ähnlich ist und keine klinisch bedeutsamen Unterschiede in Sicherheit, Reinheit oder Wirksamkeit aufweist. Die Kennzeichnung von Biosimilars muss das Produkt eindeutig als Biosimilar kennzeichnen und den Namen des Referenzprodukts enthalten. Das lila Buch listet alle zugelassenen biologischen Produkte und zugelassenen Biosimilars zur Überprüfung der Nomenklatur und des Austauschbarkeitsstatus auf. Produkte, die als austauschbar gekennzeichnet sind, können wie Generika von Apothekern ersetzt werden, wohingegen nicht austauschbare Biosimilars zuerst vom Verschreiber zugelassen werden müssen.
Die Immunogenität gehört zu den wichtigsten Sicherheitsbedenken für biologische Produkte. Patienten können Antikörper gegen Medikamente entwickeln, die möglicherweise die therapeutische Wirkung verringern oder Nebenwirkungen auslösen können. Auf dem Etikett sollten die Ergebnisse klinischer Studien zur Immunogenität sowie, soweit verfügbar, identifizierte Risikofaktoren und Empfehlungen zur Überwachung durch den Arzt enthalten sein. Da sich Biologika bei breiterer klinischer Anwendung manchmal anders verhalten als in kontrollierten Studien, verlangt die FDA häufig Sicherheitsstudien nach der Markteinführung. Die Anforderungen variieren je nach Produkt und epidemiologischem Risiko. Bei der Überarbeitung der Leitlinien für 2025 durch Health Canada werden weniger Anforderungen an präskriptive Daten eingeführt, wobei bestimmte Ausnahmen für Studien gelten, obwohl die Konsultationen zu den endgültigen Leitlinien noch nicht abgeschlossen sind. Wenn sich die Kennzeichnung auf Verpflichtungen bezieht, die nach der Markteinführung eingegangen sind, signalisiert das den Anbietern, dass sich die Sicherheitsinformationen weiterentwickeln und nicht statisch sind.
Internationale Harmonisierung der Kennzeichnung
Die globale Kennzeichnung ist nicht einheitlich. Unternehmen, die Produkte grenzüberschreitend verkaufen, sehen sich in einigen Regionen mit sich überschneidenden und in anderen widersprüchlichen Regeln konfrontiert. ICH hat viele dieser Unterschiede durch von den wichtigsten Behörden verabschiedete Richtlinien verringert, aber es gibt nach wie vor wichtige Unterschiede.
Zu den einflussreichsten ICH-Bemühungen gehört die M4-Richtlinie, mit dem ein gemeinsames Format für technische Dokumente eingeführt wurde. Anstatt für jede Aufsichtsbehörde ein neues Paket zu erstellen, können Unternehmen das ICH-Format verwenden, um frühe Forschungsergebnisse und klinische Daten einheitlich zu organisieren. Dieselbe Vorlage enthält außerdem die von den Aufsichtsbehörden erwartete Qualität der Informationen, sodass sie ohne Nacharbeit an mehrere Behörden gesendet werden können. Das ist das, was die Branche einem universellen Standard am nächsten kommt.
Regionale Erwartungen prägen immer noch, wie Etiketten in der Praxis aussehen. Die EMA verlangt beispielsweise Packungsbeilagen mit Patienteninformationen in allen EU-Sprachen, von denen jede im Rahmen des Zulassungsverfahrens geprüft und genehmigt wird. Übersetzungen können nicht einfach die englische Version wiedergeben, sie müssen den lokalen Kontext und die Gesundheitskompetenz widerspiegeln und gleichzeitig medizinisch präzise sein. Das japanische PMDA geht einen anderen Weg, dessen Beilagenanforderungen eng an die lokale klinische Praxis und die behördlichen Normen gebunden sind.
Der grenzüberschreitende Transport von Drogen erhöht die eigene Komplexität. Die Etiketten müssen den Anforderungen des Zielmarktes entsprechen, auch wenn die Rezeptur unverändert ist. Bestimmte Behörden fügen lokale Anforderungen wie Betriebslizenznummern oder Identifikatoren für die Importregistrierung hinzu. Um die Anforderungen zu erfüllen, müssen Unternehmen häufig separate Etikettendesigns erstellen, auch wenn das zugrunde liegende Produkt identisch ist.
Vorschriften zur Werbekennzeichnung
Die FDA behandelt jedes Material, das ein Hersteller über seine Produkte verteilt, als Kennzeichnung, unabhängig davon, ob es sich um eine Zeitschriftenwerbung, ein Handzettel an einem Konferenzstand oder eine Verkaufspräsentation handelt. All dies muss mit dem von der FDA zugelassenen Etikett übereinstimmen. Diese Anforderung wird durch den Grundsatz des fairen Gleichgewichts durchgesetzt: Risiken und Vorteile müssen gleich gewichtet werden. In einer Anzeige, die auf einen verringerten Krankheitsverlauf hinweist, müssen auch Nebenwirkungen, Gegenanzeigen und Nutzungsbeschränkungen gleichermaßen sichtbar sein. Risiken im Kleingedruckten zu verheimlichen oder sie in Werbespots zu beschönigen, ist eine der schnellsten Methoden, um ein Warnschreiben der FDA zu erhalten.
Das Office of Prescription Drug Promotion der FDA überwacht sowohl die formelle Werbung als auch die Verkaufsaktivitäten. Übertriebene Aussagen zur Wirksamkeit, Minimierung von Risiken, Verdrängen von Anwendungen, die nicht auf dem Etikett zugelassen sind, oder irreführende Vergleiche mit Wettbewerbern fallen in ihren Anwendungsbereich. Da die FDA die Kennzeichnung weit gefasst hat, zählen nicht nur Zeitschriftenanzeigen und Ausstellungsmaterial, sondern auch die mündlichen Aussagen, die Vertriebsmitarbeiter in Arztpraxen machen. Geringfügige Verstöße können zu Schreiben ohne Titel führen, während schwerwiegende Fälle zu Abmahnungen und einer Eskalation der Durchsetzung führen.
Digitale Kennzeichnung und elektronische Ressourcen
Elektronische Verschreibungsinformationen sind zum wichtigsten Vertriebskanal für die Arzneimittelkennzeichnung geworden. Die FDA ermöglicht es Unternehmen, Verschreibungsinformationen per E-Mail oder auf andere elektronische Weise an Gesundheitsdienstleister weiterzuleiten, die damit einverstanden sind, sie auf diese Weise zu erhalten. Dadurch wird die Verteilung auf Papier reduziert und gleichzeitig beschleunigt, wie schnell Aktualisierungen der Etikettierung die verschreibenden Ärzte erreichen.
Die strukturierte Produktkennzeichnung (SPL) betreibt die technische Infrastruktur. SPL verwendet die XML-Formatierung, um Verschreibungsinformationen in einem maschinenlesbaren Format zu kodieren. Wenn Apotheken nach Arzneimittelwechselwirkungen suchen oder elektronische Patientenakten Kontraindikationen kennzeichnen, beziehen sie Daten aus SPL-Dateien, die bei der FDA eingereicht wurden. Durch diese Automatisierung werden wichtige Informationen direkt am Behandlungsort angezeigt, ohne dass manuelle Referenzprüfungen erforderlich sind.
QR-Codes schließen die Lücke zwischen dem begrenzten Verpackungsraum und der gesamten Palette digitaler Inhalte, die Patienten und Anbieter möglicherweise benötigen. Durch das Scannen eines Codes können Verschreibungsinformationen, Arzneimittelleitfäden, Videos zur Verabreichung oder aktualisierte Sicherheitshinweise angezeigt werden. Die FDA verlangt noch keine QR-Codes, aber die Reiserichtung ist klar. Die EU hat mit Initiativen zur elektronischen Kennzeichnung bereits die Führung übernommen, und die Diskussionen in den USA über den Ersatz linearer Barcodes nehmen Fahrt auf. QR-fähige Verpackungen werden in der Branche schnell zum Standard für die Bereitstellung aktualisierter Sicherheits-, Verschreibungs- und Rückverfolgbarkeitsinformationen. Die Frage ist, wann und nicht ob sich diese Standards durchsetzen. Neue digitale Tools entstehen, obwohl die regulatorischen Rahmenbedingungen einige Zeit brauchen, um aufzuholen:
- Erweiterte Realität Anwendungen befinden sich in der Phase der explorativen Studien, wobei in einigen behördlichen Sandboxen getestet wird, ob Patienten ein Smartphone auf eine Medikamentenflasche richten können, um Visualisierungen der Wirkungsweise des Arzneimittels zu sehen oder personalisierte Dosiserinnerungen zu erhalten.
- Blockchain-Systeme werden auf manipulationssichere Aufzeichnungen von Kennzeichnungsänderungen und auf eine stärkere Überprüfung der Lieferkette getestet
- KI-gestützte Übersetzungstools könnte eines Tages eine Kennzeichnung in Echtzeit in der bevorzugten Sprache des Patienten generieren.
Bevor neue Technologien über den Pilotbetrieb hinausgehen, müssen die Aufsichtsbehörden zunächst definieren, wie sie validiert werden, und dann Regeln für die Inhaltskontrolle und Barrierefreiheit festlegen.
Klinische Bedeutung einer ordnungsgemäßen Kennzeichnung
Im Jahr 2016 Hyoscyamin-Tabletten wurden zurückgerufen wenn die Chargen Tabletten mit stark inkonsistenter Stärke enthielten — einige superpotent, andere subpotent. Patienten, die ihrer Meinung nach Standarddosen einnahmen, erhielten tatsächlich unvorhersehbare Mengen an Medikamenten. Jahre zuvor, Pfizer zurückgerufen Ungefähr 1 Million Packungen Antibabypillen, bei denen die Tabletten durch eine falsche Verpackung in der falschen Reihenfolge angeordnet wurden, was die empfängnisverhütende Wirksamkeit von Patienten beeinträchtigte, die keinen Grund zu der Annahme hatten, dass etwas nicht in Ordnung war.
Kennzeichnungsfehler wie diese sorgen für Schlagzeilen, aber eine korrekte Kennzeichnung unterstützt im Stillen jeden Tag klinische Entscheidungen. Wenn Gegenanzeigen leicht zu finden sind, können Notärzte schnell handeln, um gefährliche Arzneimittelwechselwirkungen zu vermeiden. Krankenschwestern verlassen sich aus einem anderen Grund auf Dosierungstabellen und verwenden sie, um zu bestätigen, dass die verschriebene Dosis innerhalb der zugelassenen Bereiche liegt, bevor das Medikament verabreicht wird. In Apothekensystemen tauchen deutliche Warnungen vor Arzneimittelwechselwirkungen auf, was zu Überprüfungen führt, bei denen gefährliche Kombinationen erkannt werden, bevor Medikamente die Patienten erreichen.
Einige Labels entsprechen technisch den Anforderungen, versagen aber in der Praxis immer noch. So sind beispielsweise Mindestschriftgrößen in Krankenhauskorridoren bei schwachem Licht oft schwer lesbar. Gesetzliche Formulierungen sind zwar korrekt, aber so formuliert, dass Ärzte sie in Notfällen nicht sofort erkennen. Barcodes stellen ein anderes Problem dar: Sie können zunächst die Inspektion bestehen, aber beim Drucken verschlechtern sie sich, was die automatische Überprüfung am Behandlungsort stört.

Pflichten des medizinischen Fachpersonals
Verschreibungsinformationen in einer Apothekendatenbank sind nutzlos, sofern sie nicht als Grundlage für Entscheidungen in jedem Behandlungsschritt dienen. Das bedeutet, dass Ärzte die Gegenanzeigen überprüfen, bevor sie eine Bestellung aufgeben, Apotheker die Dosis anhand der angegebenen Indikationen bestätigen und das Pflegepersonal die Durchstechflasche liest, bevor sie das Medikament zubereiten. Wenn man versteht, wie sich die Etikettierung im Prozess der Medikamenteneinnahme vollzieht, kann man erkennen, wo Sicherheitslücken bestehen. Jeder Experte verlässt sich an unterschiedlichen Entscheidungspunkten auf unterschiedliche Kennzeichnungsformate, sodass eine Kette von Überprüfungsschritten entsteht, die nur funktioniert, wenn jedes Glied stimmt. Das folgende Flussdiagramm bildet diese Kontaktpunkte ab und zeigt, wie Fehler, die an einem Checkpoint vorbeirutschen, zu dem Problem werden, das die nächste Person auffangen muss:
Dieses vernetzte System bedeutet, dass Pharmahersteller die Kennzeichnung nicht als einfache regulatorische Checkbox betrachten können. Wenn die Verschreibungsinformationen schlecht organisiert sind, übersehen Ärzte, die unter Zeitdruck arbeiten, sie möglicherweise völlig. Apotheker stoßen auf ein weiteres Problem, wenn die Terminologie auf den Kartons nicht einheitlich ist, was ihre Überprüfung verlangsamt und davon ablenkt, echte Fehler zu erkennen. Wichtige Warnungen, die in den Absätzen zur Patientenbeschriftung versteckt sind, werden von den Personen, die sie am dringendsten benötigen, nicht gelesen. Jeder Fehler bei der Etikettierung durchläuft das System und vervielfacht die Wahrscheinlichkeit, dass ein Fehler einen Patienten erreicht, bevor ihn jemand bemerkt.
Einhaltung von Kennzeichnungsvorschriften und Aktualisierungen
Wenn neue Sicherheitsinformationen vorliegen, kann die FDA gemäß Abschnitt 505 (o) (4) des FD&C Act Aktualisierungen der Kennzeichnung verlangen. Die Art der Einreichung hängt von der Änderung ab. Redaktionelle Korrekturen, die die Sicherheit oder Wirksamkeit nicht beeinträchtigen, können in Jahresberichten dokumentiert werden. Änderungen von Indikationen, Dosierungen, Gegenanzeigen oder Warnhinweisen bedürfen einer vorherigen Genehmigung durch die FDA, bevor sie wirksam werden. Bei sicherheitsrelevanten Updates geht der Prozess schnell voran:
- Das FDA-Benachrichtigungsschreiben beschreibt die neuen Sicherheitsbedenken.
- 30-tägiges Antwortfenster für das Unternehmen, um die überarbeitete Kennzeichnung einzureichen oder zu erklären, warum eine Änderung nicht erforderlich ist.
- Für Vorschläge, die die Anforderungen erfüllen, folgt eine Überprüfung und Genehmigung durch die FDA.
- Eine behördliche Anordnung kann innerhalb von 15 Tagen nach Ende der Gespräche erteilt werden, wenn das Unternehmen und die FDA keine Einigung erzielen können.
Identische Medikamente sind auf verschiedenen Märkten manchmal mit unterschiedlichen Warnhinweisen versehen. Das passiert, wenn die EMA Monate vor der FDA ein Sicherheitsupdate genehmigt, was zu Lücken in der Versionskontrolle zwischen den Ländern führt. Nachverfolgen, welche Version auf welchem Markt gilt, und dafür zu sorgen, dass die Spezifikationen an den Produktionsstandorten korrekt sind, sind nur ein Teil der Herausforderung. Unternehmen benötigen außerdem Sicherheitsvorkehrungen, um zu verhindern, dass die Bestände während des Vertriebs durcheinander geraten, insbesondere wenn mehrere Regionen betroffen sind.
Wenn Angehörige der Gesundheitsberufe Probleme bei der Kennzeichnung feststellen, können sie diese über MedWatch der FDA melden. Die Hersteller müssen untersuchen und feststellen, ob Korrekturen erforderlich sind. Das System funktioniert nur, wenn Fachleute tatsächlich melden, was sie sehen, anstatt davon auszugehen, dass es jemand anderes tun wird.
Eine nicht konforme Kennzeichnung löst eine Kette von FDA-Durchsetzungsmaßnahmen aus. Inspektoren vermerken in der Regel geringfügige Verstöße auf einer FDA-483, für die eine schriftliche Antwort und ein Korrekturplan erforderlich sind. Bei ernsteren Problemen werden Abmahnungen eingereicht, und diese Schreiben werden öffentlich bekannt. Dies wird häufig sowohl von den Aufsichtsbehörden als auch vom Markt unter die Lupe genommen. Wenn die Probleme weiterhin bestehen, kann die FDA zu einem Genehmigungsdekret eskalieren oder sogar die Produktion einstellen. Verstöße gegen die Kennzeichnung führen auch zur Haftung, wenn Patienten Schaden erleiden. Rückrufe und Lieferunterbrechungen führen zu direkten finanziellen Verlusten, während Reputationsschäden die Marktposition noch lange nach der Behebung des Compliance-Problems untergraben.
Allgemeine Probleme und Lösungen bei der Etikettierung
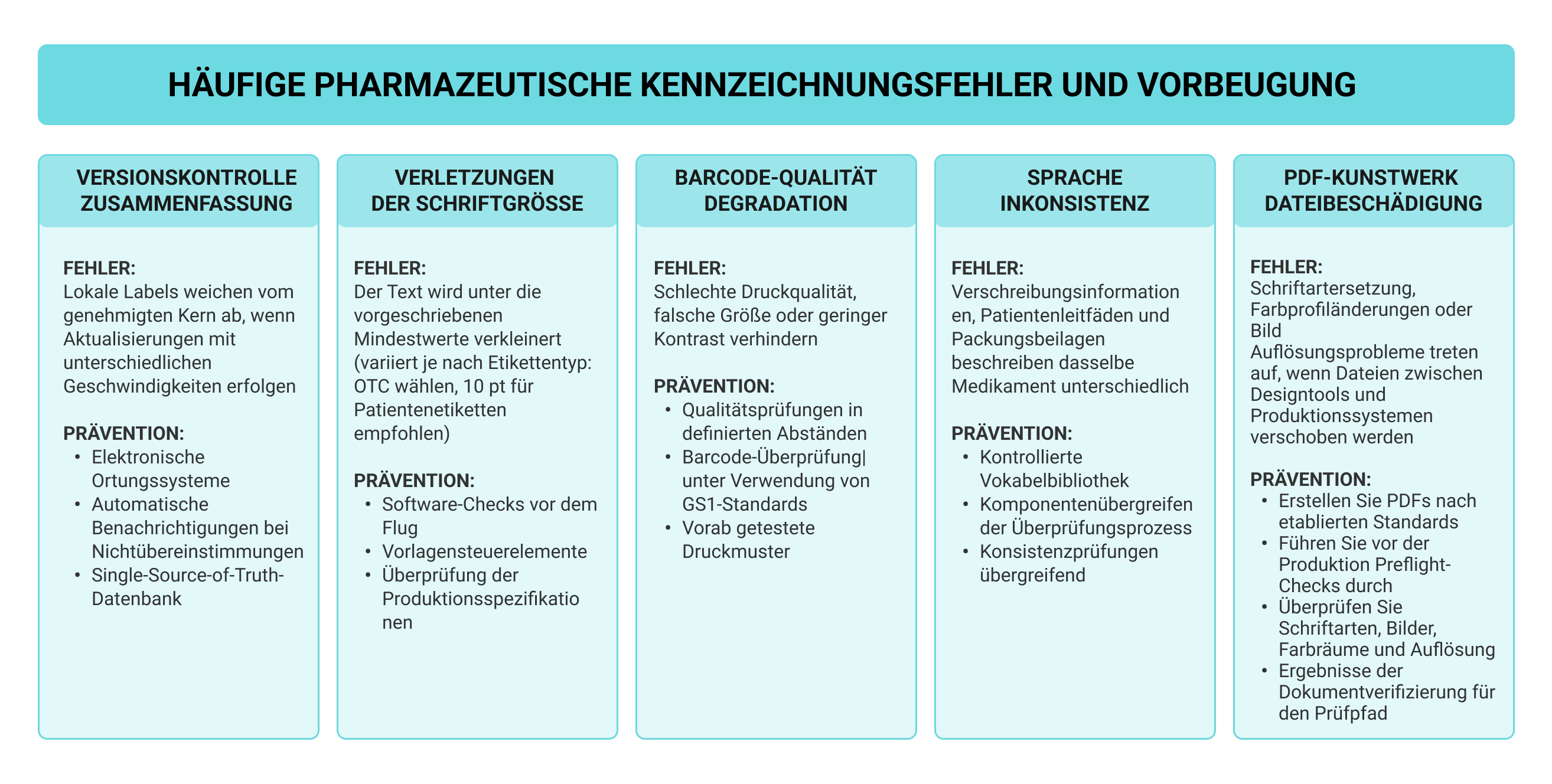
Die meisten Probleme mit der Einhaltung der Kennzeichnungsvorschriften sind darauf zurückzuführen, dass die Versionskontrolle auf den globalen Märkten ausfällt. Unternehmen, die Produkte in mehreren Regionen vermarkten, beobachten, wie lokale Etiketten von den wichtigsten Unternehmensdatenblättern abweichen, wenn Aktualisierungen mit unterschiedlichen Geschwindigkeiten erfolgen. Die Übersetzung macht das Ganze noch schlimmer, da eine genaue Anpassung an jede Sprache erforderlich ist und gleichzeitig die Konsistenz mit dem genehmigten Quellmaterial gewahrt bleibt. Eine Koordination, die die manuelle Nachverfolgung im großen Maßstab nicht bewältigen kann.
Technische Fehler werden ständig manuell überprüft. Die Schriftgrößen schrumpfen unter den Anforderungen, wenn wenig Platz zur Verfügung steht. Die Barcode-Qualität verschlechtert sich beim Drucken. Wenn Etikettengrafiken zwischen Designtools und Produktionssystemen hin und her bewegt werden, kann es bei PDF-Dateien zu Schriftwechseln kommen oder Farbprofiländerungen treten leise auf. Die Preflight-Software erkennt diese Probleme vor der Produktion, indem sie Schriften, Bilder, Farbräume und die Auflösung automatisch überprüft.

Compliance-Teams haben grundlegende Prüfungen hinter sich gelassen, indem sie automatisierte Workflows mit Preflight-Profilen in ihre Prozesse eingebettet haben. Diese Workflows decken Probleme in Echtzeit auf, sodass die Teams Fehler korrigieren können, sobald sie auftreten. Die Preflight-Ergebnisse dokumentieren jede Korrektur, bevor die Dateien in die Produktion übergehen. Teams verwenden Tools wie Adobe Acrobat, um vor dem Drucken zu überprüfen, ob die Etiketten den FDA- und ISO-PDF-Standards entsprechen. So wird ein einheitliches Erscheinungsbild und eine einheitliche Genauigkeit in allen genehmigten Versionen gewährleistet. Wenn dieser Prozess standardisiert ist, reduzieren Unternehmen den Aufwand für erneutes Drucken und kostspielige Korrekturen und stellen gleichzeitig sicher, dass wichtige Details in jeder Version korrekt bleiben, sodass die Dokumentation stets prüfungsbereit ist.
Moderne Produktionstools unterstützen auch vollständige PDF-Workflows, von der Dateierstellung in InDesign bis zur Archivierung in zentralisierten Systemen. Für Kunden, die mit Auftragsherstellern zusammenarbeiten, stellt die gemeinsame Nutzung dieser Dateien sicher, dass die endgültige Ausgabe, die die Druckmaschine erreicht, exakt den genehmigten Spezifikationen entspricht. Das Ergebnis ist eine Produktion, die für alle Anwender und Produktkategorien weltweit einheitlich bleibt. Für globale Aktivitäten gewährleistet das Speichern und Teilen von Dateien über Plattformen wie Google Drive die Versionskontrolle und ermöglicht die automatische Anzeige von Dokumenten im richtigen Format.
In Workflows zur Arzneimittelkennzeichnung bezieht sich Preflight-Software auf automatische Tools zur Dateiüberprüfung, die Probleme wie Schriftarten, Bilder, Farbräume oder Auflösung vor der Produktion kennzeichnen. Diese Prüfungen stellen die Genauigkeit und Konformität der Etikettierung von der Genehmigung bis zum endgültigen Druck sicher. Wenn Automatisierungs- und Berichtstools in die Produktionssysteme integriert sind, müssen Qualitätsteams keine Kompromisse mehr eingehen. Sie können Fehler vor dem Drucken beheben und gleichzeitig sicherstellen, dass jede Datei den FDA-Anforderungen und PDF-Industriestandards entspricht. Es gibt keine perfekte Lösung für die Einhaltung der Etikettierungsvorschriften, aber integrierte Preflight-Checks mit Automatisierung verhindern die meisten Fehler bereits vor der Produktion.
Häufig gestellte Fragen zur Einhaltung der Kennzeichnungsvorschriften:
- Woher weiß ich, ob eine Änderung einer vorherigen Genehmigung bedarf? Wenn sich eine Änderung auf Indikationen, Dosierungen, Warnhinweise oder Kontraindikationen auswirkt, muss die FDA sie vor der Umsetzung überprüfen und genehmigen. Kleinere redaktionelle Änderungen und Korrekturen können in der Regel warten und im jährlichen Update gemeldet werden. Notieren Sie immer, welche Version aktiv ist, und bewahren Sie die Dokumentation auf, aus der hervorgeht, wie mit Aktualisierungen umgegangen wurde.
- Was mache ich, wenn ich nach der Genehmigung Kennzeichnungsfehler feststelle? Fehler, die nach der Zulassung entdeckt wurden, müssen über das MedWatch-System der FDA gemeldet werden. Nach der Meldung untersucht das Qualitätsteam das Problem, identifiziert die Grundursache und führt innerhalb des festgelegten Workflows eine Lösung durch. Durch die Dokumentation, wie die Korrektur vorgenommen wurde, ist das Protokoll jederzeit zur Überprüfung bereit.
- Warum muss der National Drug Code (NDC) auf den Etiketten erscheinen? Der National Drug Code (NDC) ist die eindeutige Kennzeichnung des Produkts, die das Produkt mit der FDA-Registrierung verknüpft und Verwechselungen bei der Abgabe verhindert. In der Drucklinie liefert er die Informationen, die erforderlich sind, um zu bestätigen, dass jede Packung und jede Packung der richtigen Arzneimittelversion entspricht.
- Wer überprüft und genehmigt pharmazeutische Etiketten? In den USA ist das Center for Drug Evaluation and Research (CDER) der FDA für die Überprüfung und Zulassung von Etiketten verantwortlich. Für kontrollierte Substanzen setzt die Drug Enforcement Administration (DEA) zusätzliche Zulassungs- und Warnvorschriften durch. Anleitungen sind direkt über die Etikettierungsressourcen der FDA auf den offiziellen Webportalen der Behörde verfügbar.
- Was sind die Strafen bei Nichteinhaltung? Zu den Strafen können FDA-483-Feststellungen, Abmahnungen, Rückrufe oder die Einstellung der Produktion gehören. Diese Maßnahmen sind auch mit erheblichen finanziellen Kosten verbunden, darunter die Durchführung von Rückrufen, Produktverlust, Produktionsstopp und Kosten für Problembehebungen. Die höheren Kosten entstehen oft erst später, da Unternehmen mit Reputationsschäden und potenzieller Haftung konfrontiert sind. Sie laufen auch Gefahr, den Marktzugang zu verlieren. Die Einhaltung der Kennzeichnungsanforderungen vermeidet diese Folgen und schützt die Patienten.
- Welche Tools helfen dabei, die Einhaltung der Kennzeichnungsvorschriften sicherzustellen? Teams verlassen sich häufig auf Preflight-Software zusammen mit Tools wie Adobe Acrobat- oder InDesign-Plug-ins, um PDF-Dateien vor dem Drucken zu überprüfen. Diese Tools bestätigen, dass Schriftarten, Bilder, ICC-Profile und Farbräume korrekt sind. Sie erstellen außerdem einen Preflight-Bericht mit Einzelheiten zu jeder Korrektur und speichern jede Version, sodass Audits schnell durchgeführt werden können und die Aufzeichnungen übersichtlich bleiben.
- Können Sie ein Beispiel für häufige Fehler geben, die Software erkennt? Zu den häufigsten Problemen gehören Transparenzkonflikte, fehlende oder schlecht aufgelöste Logos und Schriftartenersetzungen in PDF-Workflows. Wenn diese Probleme erkannt werden, bevor die Dateien gedruckt werden, wird das erneute Drucken reduziert und eine gleichbleibende Qualität in allen Produktkategorien gewährleistet.
- Welche Dokumentation sollten Unternehmen führen? Unternehmen sollten ein Archiv führen, das zugelassene Labels sowie Einreichungspakete und behördliche Korrespondenz enthält. Die Dokumente müssen jederzeit zur Einsichtnahme bereit sein. Viele Organisationen bieten auch interne Schulungen an, damit die Mitarbeiter einheitliche Dokumentationspraktiken einhalten.
- Wer ist intern für die Überprüfung und Genehmigung von Arzneimitteletiketten verantwortlich? Während die FDA oder andere Aufsichtsbehörden die endgültige Genehmigung erteilen, wird der interne Überprüfungsprozess zunächst von den firmeneigenen Teams durchgeführt. Die Mitarbeiter für regulatorische Angelegenheiten und Qualitätssicherung sind in der Regel federführend und greifen auf zusätzliches Fachwissen zurück, wenn medizinische Genauigkeit, rechtliche Risiken oder das Verpackungsdesign genauer geprüft werden müssen. Jeder Schritt wird dokumentiert, sodass das Unternehmen bei der Einreichung eines Etiketts bei der FDA nachweisen kann, dass alle internen Kontrollpunkte abgeschlossen wurden.
Wichtige Ressourcen:
- FDAs“Sicherheitsüberlegungen für das Design von Behälteretiketten und Kartonetiketten zur Minimierung von Medikationsfehlern„(Mai 2022)
- „Kennzeichnung verschreibungspflichtiger Humanarzneimittel und biologischer Produkte — Umsetzung der PLR-Anforderungen an Inhalt und Format„
- FDA Abteilung für Prävention und Analyse von Medikationsfehlern (für spezielle Compliance-Fragen)
Künftige Richtungen der Arzneimittelkennzeichnung
Die meisten klinischen Einrichtungen sind nicht mehr auf Verschreibungsinformationen in Papierform angewiesen. Stattdessen elektronischer Vertrieb liefert strukturierte Produktkennzeichnungsdaten, die Apothekensysteme verwenden, um Warnmeldungen zu Arzneimittelwechselwirkungen zu generieren und klinische Entscheidungen zu unterstützen. QR-Codes verbinden physische Verpackungen mit digitalen Inhalten, die nicht auf Behälter passen. Dies sind keine neuen Technologien, sie sind die Realität, wie Etikettierung heute funktioniert.
Weltweite Harmonisierung through ICH hat die wichtigsten Dokumentationsanforderungen aufeinander abgestimmt, aber die regionalen Unterschiede sind nach wie vor tief verwurzelt. Zwar zeigen Bemühungen wie die Annäherung des kanadischen XML-PM-Formats an die SPL der FDA Fortschritte, doch die Unterschiede zwischen den Anforderungen an die Packungsbeilage und den Leitlinien für Biologika sind von Land zu Land nach wie vor erheblich. Die EMA verlangt Patienteninformationsbroschüren in allen EU-Amtssprachen mit marktspezifischen Anpassungen. Die japanische PMDA hat eigene Anforderungen an die Packungsbeilage. Unternehmen, die Produkte marktübergreifend verwalten, verarbeiten immer noch mehrere Etikettenversionen für identische Medikamente, obwohl bessere Datensysteme die Fehler reduziert haben, die durch die manuelle Nachverfolgung entstanden sind.
Barrierefreiheit für Patienten ist ein Bereich des ständigen Wandels. Die Regulierungsbehörden gehen über allgemeine Aussagen über einfache Formulierungen hinaus und definieren nun klarer, was als patientenfreundliche Kommunikation gilt, und nicht, was lediglich eine Checkbox zur Einhaltung der Vorschriften erfüllt. Digitale Formate öffnen die Tür zur Personalisierung. Ein Patient könnte die Schriftgröße aus Gründen der Lesbarkeit anpassen, zu seiner bevorzugten Sprache wechseln oder eine Audioversion wählen. Diese Tools sind noch nicht offiziell für behördliche Zwecke anerkannt. Sowohl die US-amerikanischen als auch die kanadischen Behörden führen derzeit gesetzliche und regulatorische Überprüfungen durch, was bedeutet, dass die adaptive Kennzeichnung eher ein sich entwickelnder Standard als eine etablierte Praxis ist.
Gleichzeitig stützen sich die Aufsichtsbehörden stärker auf Beweise aus der realen Welt. Daten aus elektronischen Patientenakten und Patientenregistern spielen heute eine wachsende Rolle bei der Gestaltung von Kennzeichnungsanforderungen und Zulassungsentscheidungen. Da Behörden reale Daten in die Sicherheitsüberwachung einbeziehen, kann es vorkommen, dass Aktualisierungen der Etiketten schneller erfolgen als im herkömmlichen jährlichen Zyklus. Der eigentliche Test wird darin bestehen, wie schnell die Hersteller ihre Systeme anpassen, da häufige Änderungen das Risiko von Fehlern bei der Versionskontrolle und Etikettierung erhöhen.
Verify von GlobalVision wurde für die Inspektionsherausforderungen entwickelt, mit denen Pharmateams konfrontiert sind, wenn die Einhaltung der Vorschriften nicht darauf warten kann, dass menschliche Fehler auftauchen. Erfahren Sie, wie Verify funktioniert.
