

Même un seul chiffre transposé sur une étiquette pharmaceutique peut mettre les patients en danger et entraîner des rappels coûteux. Les entreprises qui se tiennent prêtes aux inspections sont celles qui connaissent non seulement les exigences, mais qui savent également où les erreurs se produisent le plus souvent.
Introduction à l'étiquetage des produits pharmaceutiques
L'étiquetage des produits pharmaceutiques inclut tous les éléments figurant sur l'emballage des médicaments. Cela couvre non seulement les mots, mais également les éléments graphiques tels que les logos, les bandes de couleur, les codes-barres, le braille et les symboles réglementaires, allant du texte du flacon aux informations posologiques en passant par le matériel promotionnel et les notices d'emballage. La définition de la FDA selon la Loi fédérale sur les aliments, les médicaments et les cosmétiques jette un large filet : s'il accompagne votre médicament, c'est son étiquetage. Mais les différents marchés sont régis par des régulateurs différents. Aux États-Unis, il est géré par le Center for Drug Evaluation and Research de la FDA. L'Europe confie cette tâche à l'EMA, tandis que le Japon s'appuie sur le PMDA. Le Conseil international pour l'harmonisation (ICH) a réduit les écarts grâce à l'harmonisation, mais une véritable harmonisation du contenu reste un travail en cours. Les différences régionales persistent, non seulement en termes de langue et de traduction, mais aussi en ce qui concerne les exigences en matière de données, l'ordre des contenus et les priorités locales en matière de risques/avantages. Cela signifie qu'une étiquette qui approuve l'examen de la FDA peut encore avoir besoin d'être révisée avant que les régulateurs européens ne l'acceptent.
L'étiquetage des produits pharmaceutiques couvre également les médicaments en vente libre (OTC), qui ont leur propre format d'étiquette standardisé « Drug Facts ». Contrairement à l'étiquetage des médicaments sur ordonnance, qui est destiné aux prestataires de soins de santé, l'étiquetage en vente libre doit être explicite pour les consommateurs. Les règles de typographie pour l'étiquetage OTC sont exceptionnellement strictes. Le règlement de la FDA sur l'information sur les médicaments exige que le texte soit composé d'au moins 6 points, avec des titres en gras et une mise en page qui reste lisible à bout de bras. Dans ce format, la FDA spécifie également les éléments qui doivent apparaître, dans cet ordre :
- Ingrédient (s) actif (s) : nom et concentration par unité posologique
- Objectif : la catégorie de médicament ou l'effet escompté
- Utilisations : indications approuvées
- Avertissements : y compris les mentions « Ne pas utiliser », les précautions à prendre en cas de grossesse/d'allaitement et les cas où il convient de consulter un médecin
- Directions : instructions posologiques pour une utilisation en toute sécurité
- Ingrédients inactifs : répertorié pour la sensibilisation aux allergies
Des instructions posologiques peu claires et des contre-indications mal placées laissent les consommateurs sans les informations complètes dont ils ont besoin pour utiliser les produits en toute sécurité. Le contrôle réglementaire ne cesse de s'intensifier car les risques de malentendu sont très élevés.
Ressources d'étiquetage de la FDA pour les médicaments sur ordonnance
La FDA gère plusieurs bases de données pour accéder aux informations d'étiquetage officielles. Drogues @FDA est le plus complet, contenant les informations d'approbation, les détails de prescription et l'historique de l'étiquetage de milliers de médicaments approuvés. Vous voulez voir comment un label a évolué ? C'est ici que vous effectuez votre recherche.
Daily Med, géré par la National Library of Medicine avec la FDA, publie un étiquetage standard des produits dans un format structuré. Les professionnels de santé l'utilisent lorsqu'ils vérifient les informations de prescription sur le lieu des soins. Lorsque vous naviguez sur l'un ou l'autre des portails, commencez par la section « Points forts », qui résume les informations de prescription essentielles avant les sections détaillées sur la pharmacologie clinique et les exigences de stockage.
Lors des inspections réglementaires, les auditeurs comparent les étiquettes de production aux soumissions officielles. La version approuvée par la FDA représente ce que l'agence a examiné et approuvé, tandis que l'étiquetage actuel reflète les mises à jour les plus récentes. Les équipes de qualité doivent se référer à la version approuvée par la FDA comme référence de conformité, et non à ce qui est publié sur DailyMed en ce moment. Cette distinction est importante lorsque les inspecteurs se présentent avec vos documents de soumission en main.
Composantes d'information posologique
Les informations de prescription professionnelles suivent le format des règles d'étiquetage des médecins, qui divise le contenu en deux sections principales : points forts et informations posologiques complètes.


L'étiquetage destiné aux patients pédiatriques nécessite des considérations spéciales au-delà de ce qui apparaît dans la section 8 des informations posologiques. En vertu de la loi sur les meilleurs produits pharmaceutiques pour enfants (BPCA) et de la loi sur l'équité en matière de recherche pédiatrique (PREA), les fabricants doivent soumettre des données cliniques spécifiques à la pédiatrie plutôt que de se fier uniquement à des études sur des adultes. Dans la pratique, l'étiquetage pédiatrique combine des tableaux posologiques basés sur le poids avec des instructions claires sur les formulations adaptées à l'âge. Il émet également des avertissements de sécurité lorsque les risques dépassent les avantages potentiels. Ces informations aident à prévenir les utilisations non indiquées sur l'étiquette, l'une des principales causes d'erreurs posologiques chez les enfants, en fournissant aux professionnels de santé des informations qu'ils peuvent appliquer directement dans le cadre de soins réels.
En ce qui concerne les données relatives aux essais cliniques, les informations de prescription doivent aller au-delà des résultats principaux. Pour que l'étiquetage soit significatif, il doit présenter la réduction du risque absolu d'une manière qui reflète les caractéristiques de la population étudiée et la durée du suivi. Sans ce contexte, les prestataires ne peuvent pas évaluer si les résultats des essais s'appliquent à leurs patients. En milieu hospitalier, les systèmes d'aide à la décision clinique extraient ces données pour signaler les problèmes potentiels avant que les médicaments n'atteignent les patients.
Exigences relatives à l'étiquetage des patients
L'étiquetage des patients permet de prendre des informations médicales et de les rendre accessibles aux personnes sans formation clinique. La FDA exige des formats spécifiques en fonction de la classe de médicaments et du profil de risque : guides de médicaments, notices destinées aux patients et instructions d'utilisation. Les guides de médication sont obligatoires lorsque des informations sur la prévention des effets indésirables graves sont essentielles pour une utilisation sûre. Ces documents d'une page expliquent les risques en langage clair, souvent sous forme de questions-réponses. La FDA pré-approuve le libellé exact, ce qui signifie que les fabricants ne peuvent pas modifier le contenu sans l'autorisation de l'agence.
Une communication claire en matière d'étiquetage va au-delà des simples choix de vocabulaire. La FDA s'attend à un langage clair : une voix active sur des constructions passives, des phrases courtes plutôt que des phrases complexes, une terminologie courante plutôt que du jargon médical. Lorsque des termes techniques sont inévitables, des définitions en langage clair doivent suivre immédiatement. La recherche montre que les patients sont plus susceptibles de suivre les instructions posologiques et de reconnaître les signes avant-coureurs lorsque l'étiquetage parle leur langue.
La mise en page joue un rôle tout aussi important dans la compréhension du patient. Les pages qui semblent ouvertes et structurées sont plus faciles à parcourir, et des titres solides guident les lecteurs vers les détails dont ils ont besoin. Pour les personnes âgées, un caractère plus gros fait souvent la différence entre des instructions claires et des doses oubliées. Pour renforcer cela, la FDA impose des tailles de police minimales et un contraste élevé entre le texte et l'arrière-plan, reconnaissant que l'étiquetage ne fonctionne que si les patients peuvent réellement le lire. De nombreuses équipes utilisent désormais outils de vérification automatique de la taille des polices pour garantir la conformité avant que l'emballage n'atteigne la production.
Normes d'étiquetage des cartons et des contenants
La première couche d'emballage est le contenant qui touche le médicament lui-même. Il doit contenir les informations nécessaires à une utilisation en toute sécurité. Une deuxième couche, par exemple les cartons ou les expéditeurs, soutient la chaîne d'approvisionnement en affichant des informations supplémentaires.
La FDA spécifie le contenu requis à la fois pour :
Contenant principal :
- Nom du médicament
- Force
- Voie d'administration
- Numéro de lot
- Date d'expiration
Emballage secondaire :
- Nom complet du produit
- Ingrédients actifs
- Forme posologique
- Quantité nette
- Conditions de stockage
- Informations sur le fabricant
- Code national des médicaments (NDC)
Les contraintes d'espace sur les petits conteneurs constituent un défi pratique. Lorsque le contenant est trop petit pour contenir tous les éléments requis, la FDA autorise certains détails à apparaître uniquement sur le carton, à condition que les deux restent ensemble pendant la distribution. Le Code national des médicaments (NDC) est essentiel à cet égard, car il sert d'identifiant unique reliant l'emballage à l'enregistrement auprès de la FDA. Cela réduit le risque de confusion entre les médicaments en pharmacie et en milieu clinique.
Les informations de sécurité ne sont pas présentées de manière uniforme sur tous les produits. Les conditions de stockage, telles que la réfrigération ou la protection contre la lumière, sont mises en évidence lorsque les directives spécifiques au produit l'exigent. Les substances contrôlées sont soumises à leurs propres règles : la DEA exige que le symbole du calendrier (I—V) apparaisse bien en évidence sur l'étiquette, en caractères plus grands que le nom du médicament. Si le symbole reste visible à travers l'emballage, il n'est pas nécessaire de le transporter séparément dans les emballages extérieurs. Le code-barres répond à un autre niveau de risque en permettant la traçabilité tout au long de la chaîne d'approvisionnement et en renforçant les exigences de la FDA et de la DEA.
Les exigences en matière d'étiquetage des codes-barres ont évolué parallèlement aux systèmes électroniques de médicaments. En vertu du 21 CFR 201.25, la FDA exige que de nombreuses étiquettes de médicaments sur ordonnance incluent un code-barres qui code au moins le NDC. Dans la pratique, les systèmes pharmaceutiques et cliniques scannent ce code-barres pendant la distribution ou l'administration pour confirmer le produit et détecter les incohérences que la transcription manuelle pourrait ne pas détecter.
[Visuel : boîte pharmaceutique annotée montrant l'emplacement du NDC, les spécifications du code-barres, la position de la déclaration d'avertissement, le formatage du lot/de la date d'expiration, l'affichage des conditions de stockage]
Considérations relatives à l'étiquetage des médicaments génériques
L'étiquetage des médicaments génériques suit une norme d'uniformité avec des exceptions spécifiques. La FDA exige que les médicaments génériques correspondent au médicament de référence sur les points essentiels : indications, posologie, voie d'administration, concentration et principe actif.
Le contenu clinique d'une étiquette générique doit être identique à celui du médicament répertorié de référence (RLD). Cela inclut les mises en garde et les précautions, ainsi que le libellé concernant les effets indésirables et les interactions médicamenteuses. Si la marque comporte une mise en garde encadrée, telle qu'une mise en garde concernant le risque cardiovasculaire, le générique répète la mise en garde au même emplacement et au même format. Les domaines dans lesquels les génériques ont une marge de manœuvre sont liés aux entreprises. Un fabricant peut utiliser son propre nom et ses coordonnées, mettre à jour la section « Mode de fourniture » en fonction de son emballage et omettre les déclarations de brevet ou d'exclusivité qui ne s'appliquent qu'au produit de marque.
La FDA continue de souligner la nécessité pour les génériques de maintenir l'alignement de l'étiquetage sur les informations de sécurité actuelles. En vertu de l'article 505 (o) (4), l'agence peut exiger des mises à jour lorsque de nouveaux risques sont identifiés, en veillant à ce que le médicament répertorié de référence et les génériques approuvés portent les mêmes avertissements. Alors que la surveillance de la FDA se renforce, le mécanisme d'alignement rapide en matière de sécurité a fait l'objet de réformes et le calendrier des mises à jour peut varier dans la pratique. Certains alignements sont toujours à la traîne malgré l'importance accordée à la réglementation.
Exigences en matière d'étiquetage des produits biologiques
Les produits biologiques diffèrent des médicaments traditionnels de manière importante. Les médicaments traditionnels sont synthétisés par des procédés chimiques prévisibles. Les produits biologiques sont produits dans des systèmes vivants (cellules bactériennes, levures, cultures de cellules de mammifères), où les caractéristiques du produit peuvent varier d'un lot de production à l'autre de manière à affecter la sécurité et l'efficacité.
Un biosimilaire est un produit biologique très similaire à un produit de référence déjà approuvé, sans aucune différence cliniquement significative en termes de sécurité, de pureté ou d'activité. L'étiquetage des produits biosimilaires doit clairement identifier le produit comme biosimilaire et inclure le nom du produit de référence. Le livre violet répertorie tous les produits biologiques homologués et les biosimilaires approuvés pour vérifier la nomenclature et le statut d'interchangeabilité. Les produits désignés comme interchangeables peuvent être remplacés par les pharmaciens, comme les médicaments génériques, tandis que les médicaments biosimilaires non interchangeables doivent d'abord être approuvés par le prescripteur.
L'immunogénicité est l'une des principales préoccupations en matière de sécurité des produits biologiques. Les patients peuvent développer des anticorps antimédicamenteux susceptibles de réduire l'effet thérapeutique ou de déclencher des effets indésirables. L'étiquetage doit indiquer les résultats d'immunogénicité issus des études cliniques et, le cas échéant, inclure les facteurs de risque identifiés et les suggestions de surveillance pour les cliniciens. Étant donné que les produits biologiques se comportent parfois différemment dans le cadre d'une utilisation clinique plus large que dans le cadre d'essais contrôlés, la FDA exige souvent des études de sécurité après la commercialisation. Les exigences varient en fonction du produit spécifique et du risque épidémiologique. Les révisions des lignes directrices de 2025 de Santé Canada évoluent vers des exigences de données moins prescriptives avec certaines dérogations pour les études, bien que les consultations sur les directives finales soient en cours. Lorsque l'étiquetage fait référence à des engagements après la commercialisation, il indique aux fournisseurs que les informations de sécurité évoluent et ne sont pas statiques.
Harmonisation internationale de l'étiquetage
L'étiquetage mondial n'est pas uniforme. Les entreprises qui vendent des produits au-delà des frontières sont confrontées à des règles qui se chevauchent dans certaines régions et à des règles contradictoires dans d'autres. L'ICH a réduit de nombreuses lacunes grâce à des directives adoptées par les principales autorités, mais d'importantes différences persistent.
Parmi les initiatives les plus influentes de l'ICH, citons Directive M4, qui a introduit un format de document technique commun. Plutôt que de créer un nouveau package pour chaque organisme de réglementation, les entreprises peuvent utiliser le format ICH pour organiser les premières recherches et les données cliniques de manière cohérente. La même soumission contient également les informations de qualité attendues par les régulateurs, ce qui permet de l'envoyer à plusieurs agences sans retouche. C'est ce qui se rapproche le plus d'une norme universelle dans l'industrie.
Les attentes régionales façonnent toujours l'apparence des étiquettes dans la pratique. L'EMA, par exemple, exige des brochures d'information destinées aux patients dans toutes les langues de l'UE, chacune étant examinée et approuvée dans le cadre du processus d'autorisation. Les traductions ne doivent pas simplement refléter la version anglaise, elles doivent refléter le contexte local et les connaissances en matière de santé tout en restant précises sur le plan médical. Le PMDA japonais emprunte une autre voie, avec des exigences d'insertion étroitement liées à la pratique clinique et aux normes réglementaires locales.
Le transport de drogues au-delà des frontières ajoute de la complexité en soi. Les étiquettes doivent correspondre aux exigences du marché de destination, même lorsque la formulation est inchangée. Certaines autorités ajoutent des exigences locales telles que les numéros de licence d'établissement ou les identifiants d'enregistrement des importations. Pour s'y conformer, les entreprises doivent souvent produire des modèles d'étiquettes distincts, même lorsque le produit sous-jacent est identique.
Réglementation sur l'étiquetage promotionnel
La FDA considère tout matériel distribué par un fabricant à propos de ses produits comme un étiquetage, qu'il s'agisse d'une publicité dans un journal, d'un document de cabine de conférence ou d'une présentation commerciale. Tout cela doit rester conforme à l'étiquette approuvée par la FDA. Cette exigence est appliquée selon le principe du juste équilibre : les risques et les avantages doivent être présentés avec le même poids. Une publicité qui met en évidence une progression réduite de la maladie doit également donner la même visibilité aux effets secondaires, aux contre-indications et aux limites d'utilisation. L'un des moyens les plus rapides de rédiger une lettre d'avertissement de la FDA consiste à enterrer les risques en petits caractères ou à les masquer dans des spots télévisés.
Le Bureau de promotion des médicaments sur ordonnance de la FDA surveille à la fois la publicité officielle et les activités de vente. Surestimer l'efficacité, minimiser les risques, promouvoir les utilisations non indiquées sur l'étiquette ou faire des comparaisons trompeuses avec des concurrents entrent tous dans son champ d'application. Étant donné que la FDA définit l'étiquetage de manière large, non seulement les publicités dans les journaux et le matériel des stands comptent, mais aussi les déclarations verbales faites par les représentants commerciaux dans les cabinets médicaux. Les infractions mineures peuvent donner lieu à des lettres sans titre, tandis que les cas graves entraînent des lettres d'avertissement et une intensification de l'application de la loi.
Étiquetage numérique et ressources électroniques
Les informations de prescription électroniques sont devenues le principal canal de distribution pour l'étiquetage des médicaments. La FDA permet aux entreprises de fournir des informations de prescription par e-mail ou par d'autres moyens électroniques aux prestataires de santé qui acceptent de les recevoir de cette manière, réduisant ainsi la distribution papier tout en accélérant la rapidité avec laquelle les mises à jour de l'étiquetage parviennent aux prescripteurs.
L'étiquetage structuré des produits (SPL) gère l'infrastructure technique. SPL utilise le formatage XML pour coder les informations de prescription dans un format lisible par machine. Lorsque les pharmacies vérifient les interactions médicamenteuses ou que les dossiers médicaux électroniques signalent des contre-indications, elles extraient les données des fichiers SPL soumis à la FDA. Cette automatisation affiche les informations critiques au point de service sans nécessiter de vérifications manuelles des références.
Les codes QR permettent de combler le fossé entre l'espace d'emballage limité et la gamme complète de contenus numériques dont les patients et les prestataires peuvent avoir besoin. La lecture d'un code permet d'obtenir des informations de prescription, des guides sur les médicaments, des vidéos d'administration ou des consignes de sécurité mises à jour. La FDA n'exige pas encore de codes QR, mais la direction à suivre est claire. L'UE a déjà pris l'initiative en matière d'étiquetage électronique, et les discussions américaines sur le remplacement des codes-barres linéaires s'accélèrent. Les emballages compatibles QR sont en passe de devenir la solution par défaut du secteur pour fournir des informations actualisées en matière de sécurité, de prescription et de traçabilité. Il s'agit de savoir quand, et non si, ces normes seront appliquées. De nouveaux outils numériques apparaissent alors même que les cadres réglementaires mettent du temps à rattraper leur retard :
- Réalité augmentée applications sont en phase d'essais exploratoires, certains bacs à sable réglementaires visant à déterminer si les patients pouvaient pointer un smartphone vers un flacon de médicament pour visualiser le fonctionnement du médicament ou recevoir des rappels posologiques personnalisés.
- Systèmes de chaînes de blocs sont en cours de test pour détecter toute falsification des modifications d'étiquetage et pour renforcer la vérification de la chaîne d'approvisionnement
- Outils de traduction alimentés par l'IA pourrait un jour générer un étiquetage en temps réel dans la langue préférée du patient.
Avant que les nouvelles technologies puissent aller au-delà de l'utilisation pilote, les régulateurs doivent d'abord définir la manière dont elles seront validées, puis définir des règles pour le contrôle du contenu et l'accessibilité.
Importance clinique d'un étiquetage approprié
En 2016, les comprimés d'hyoscyamine ont été rappelés lorsque les lots contenaient des comprimés d'une force extrêmement incohérente, certains superpuissants, d'autres subpuissants. Les patients qui prenaient ce qu'ils croyaient être des doses standard recevaient en fait des quantités imprévisibles de médicaments. Des années plus tôt, Pfizer rappelé environ 1 million de boîtes de pilules contraceptives lorsqu'un emballage incorrect a mis les comprimés dans le mauvais ordre, compromettant ainsi l'efficacité de la contraception pour les patientes qui n'avaient aucune raison de penser que quelque chose n'allait pas.
De tels échecs en matière d'étiquetage font la une des journaux, mais un étiquetage approprié soutient discrètement les décisions cliniques au quotidien. Lorsque les contre-indications sont faciles à détecter, les médecins urgentistes peuvent agir rapidement pour éviter les interactions médicamenteuses dangereuses. Les infirmières se fient aux tableaux posologiques pour une autre raison, les utilisant pour confirmer que la dose prescrite se situe dans les fourchettes approuvées avant l'administration du médicament. Des mises en garde importantes concernant les interactions médicamenteuses apparaissent dans les systèmes pharmaceutiques, déclenchant des examens qui détectent les combinaisons dangereuses avant que les médicaments n'atteignent les patients.
Certains labels sont techniquement conformes mais échouent toujours dans la pratique. Les tailles de police minimales, par exemple, deviennent souvent difficiles à lire en cas de faible éclairage dans les couloirs des hôpitaux. Le libellé de la réglementation peut être précis mais formulé de manière à ce que les cliniciens ne puissent pas immédiatement reconnaître en cas d'urgence. Les codes-barres présentent un problème différent : ils peuvent réussir l'inspection au début, mais se dégrader lors de l'impression, ce qui perturbe la vérification automatique sur le lieu de service.

Responsabilités des professionnels de santé
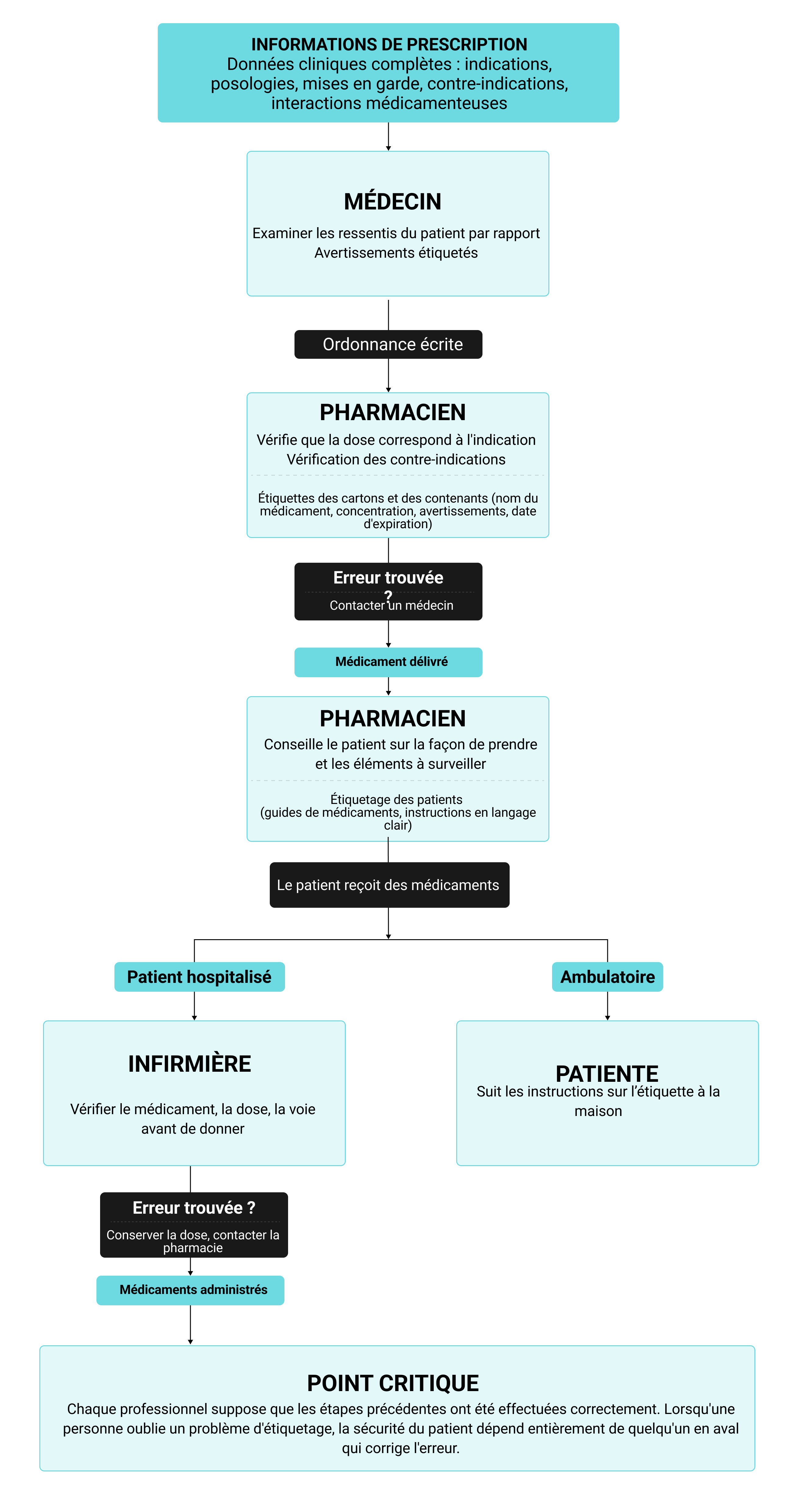
Les informations de prescription contenues dans une base de données pharmaceutiques sont inutiles à moins qu'elles ne guident les décisions à chaque étape des soins. Cela signifie que les médecins vérifient les contre-indications avant de rédiger une commande, que les pharmaciens confirment la dose par rapport aux indications indiquées sur l'étiquette et que les infirmières lisent le flacon avant de préparer le médicament. Comprendre comment l'étiquetage évolue tout au long du processus d'utilisation des médicaments révèle les vulnérabilités. Chaque professionnel s'appuie sur différents formats d'étiquetage à différents points de décision, créant ainsi une chaîne d'étapes de vérification qui ne fonctionne que lorsque chaque maillon est solide. L'organigramme ci-dessous cartographie ces points de contact et montre comment les erreurs qui passent au-delà d'un point de contrôle deviennent le problème à détecter pour la personne suivante :

Ce système interconnecté signifie que les fabricants de produits pharmaceutiques ne peuvent pas considérer l'étiquetage comme une simple case à cocher réglementaire. Lorsque l'information posologique est mal organisée, les médecins qui travaillent dans des délais serrés peuvent l'oublier complètement. Les pharmaciens rencontrent un autre problème lorsque la terminologie utilisée sur les emballages n'est pas cohérente, ce qui ralentit leur examen et les empêche de détecter les véritables erreurs. Les avertissements critiques enfouis dans de denses paragraphes d'étiquetage destinés aux patients ne sont pas lus par les personnes qui en ont le plus besoin. Chaque échec d'étiquetage se répercute sur le système, multipliant ainsi les chances qu'une erreur parvienne au patient avant que quelqu'un ne la détecte.
Conformité et mises à jour des étiquettes
Lorsque de nouvelles informations de sécurité apparaissent, la FDA peut exiger des mises à jour de l'étiquetage en vertu de l'article 505 (o) (4) de la loi FD&C. Le type de soumission dépend de la modification. Les corrections rédactionnelles qui n'affectent pas la sécurité ou l'efficacité peuvent être documentées dans les rapports annuels. Les modifications des indications, de la posologie, des contre-indications ou des mises en garde nécessitent l'approbation préalable de la FDA avant d'entrer en vigueur. Pour les mises à jour axées sur la sécurité, le processus est rapide :
- La lettre de notification de la FDA décrit le nouveau problème de sécurité.
- Délai de réponse de 30 jours permettant à l'entreprise de soumettre un étiquetage révisé ou d'expliquer pourquoi aucune modification n'est nécessaire.
- L'examen et l'approbation de la FDA suivent pour les propositions qui répondent à l'exigence.
- L'ordre de l'agence peut être émis dans les 15 jours suivant la fin des discussions si l'entreprise et la FDA ne parviennent pas à un accord.
Des médicaments identiques comportent parfois des mises en garde différentes selon les marchés. Cela se produit lorsque l'EMA approuve une mise à jour de sécurité des mois avant la FDA, ce qui crée des lacunes dans le contrôle des versions entre les juridictions. Le suivi de la version applicable à chaque marché et le maintien de spécifications précises sur les sites de production ne constituent qu'une partie du défi. Les entreprises ont également besoin de garanties pour éviter que les stocks ne soient mélangés lors de la distribution, en particulier lorsque plusieurs régions sont concernées.
Lorsque les professionnels de santé constatent des problèmes d'étiquetage, ils peuvent les signaler à la FDA via MedWatch. Les fabricants doivent étudier et déterminer si des corrections sont nécessaires. Le système ne fonctionne que lorsque les professionnels signalent réellement ce qu'ils voient au lieu de supposer que quelqu'un d'autre le fera.
Un étiquetage non conforme déclenche une série de mesures coercitives de la FDA. Les inspecteurs constatent généralement des violations mineures sur un FDA-483, ce qui nécessite une réponse écrite et un plan correctif. Les problèmes les plus graves font l'objet de lettres d'avertissement, qui deviennent publiques. Cela fait souvent l'objet d'un examen minutieux de la part des régulateurs et du marché. Si les problèmes persistent, la FDA peut passer à un décret d'approbation ou même arrêter la fabrication. Les violations de l'étiquetage entraînent également une responsabilité lorsque les patients subissent un préjudice. Les rappels et les interruptions d'approvisionnement entraînent des pertes financières directes, tandis que l'atteinte à la réputation érode la position sur le marché longtemps après la correction du problème de conformité.
Problèmes d'étiquetage courants et solutions
La plupart des problèmes de conformité en matière d'étiquetage sont dus à la défaillance du contrôle des versions sur les marchés mondiaux. Les entreprises qui commercialisent des produits dans plusieurs régions voient les étiquettes locales s'éloigner des fiches techniques de base de l'entreprise lorsque les mises à jour se produisent à des vitesses différentes. La traduction ne fait qu'empirer les choses, car elle nécessite une adaptation précise à chaque langue tout en préservant la cohérence avec les sources approuvées, exige une coordination que le suivi manuel ne peut pas gérer à grande échelle.
Les erreurs techniques font constamment l'objet d'une révision manuelle. Les tailles de police diminuent en dessous des exigences en raison de la pression de l'espace. La qualité des codes-barres se dégrade pendant l'impression. Lorsque les illustrations d'étiquettes passent des outils de conception aux systèmes de production, les fichiers PDF peuvent comporter des substitutions de police ou des changements de profil de couleur se produire discrètement. Le logiciel Preflight détecte ces problèmes avant la production en vérifiant automatiquement les polices, les images, les espaces colorimétriques et la résolution.
Les équipes de conformité ont dépassé les contrôles de base en intégrant des flux de travail automatisés dans leurs processus avec des profils de contrôle. Ces flux de travail font apparaître les problèmes en temps réel, permettant aux équipes de corriger les erreurs au fur et à mesure qu'elles apparaissent, et les résultats en avant-vol documentent chaque correction avant que les fichiers ne passent en production. Les équipes utilisent des outils tels qu'Adobe Acrobat pour vérifier que les illustrations d'étiquetage sont conformes aux normes PDF de la FDA et ISO avant l'impression, garantissant ainsi une apparence et une précision homogènes sur chaque version approuvée. Lorsque ce processus est standardisé, les entreprises réduisent les réimpressions et les corrections coûteuses tout en veillant à ce que les détails importants restent exacts dans chaque version, en gardant la documentation toujours prête à être auditée.
Les outils de production modernes prennent également en charge des flux de travail PDF complets, de la création de fichiers dans InDesign à l'archivage dans des systèmes centralisés. Pour les clients travaillant avec des fabricants sous contrat, le partage de ces fichiers garantit que le résultat final qui parvient à la presse correspond exactement aux spécifications approuvées. Le résultat est un résultat qui reste cohérent entre les utilisateurs et les catégories de produits du monde entier. Pour les opérations mondiales, le stockage et le partage de fichiers via des plateformes telles que Google Drive garantissent le contrôle des versions et permettent d'afficher automatiquement les documents dans le format approprié.
Dans les flux de travail d'étiquetage des produits pharmaceutiques, le logiciel de contrôle préalable fait référence à des outils automatisés de vérification des fichiers qui signalent des problèmes tels que les polices, les images, les espaces colorimétriques ou la résolution avant la production. Ces contrôles garantissent la précision et la conformité de l'étiquetage, de l'approbation à l'impression finale. Lorsque les outils d'automatisation et de reporting sont intégrés aux systèmes de production, les équipes de qualité ne sont plus confrontées à des compromis. Ils peuvent résoudre les erreurs avant l'impression tout en veillant à ce que chaque fichier soit conforme aux exigences de la FDA et aux normes PDF de l'industrie. Il n'existe pas de solution parfaite pour la conformité de l'étiquetage, mais les contrôles avant contrôle intégrés avec automatisation permettent d'éviter la plupart des erreurs avant la production.
Questions fréquentes concernant la conformité de l'étiquetage :
- Comment savoir si une modification nécessite une approbation préalable ? Si un changement affecte les indications, la posologie, les mises en garde ou les contre-indications, la FDA doit l'examiner et l'approuver avant sa mise en œuvre. Les modifications et corrections rédactionnelles mineures peuvent généralement attendre et être signalées dans la mise à jour annuelle. Enregistrez toujours la version active et conservez la documentation pour montrer comment les mises à jour ont été gérées.
- Que dois-je faire si je constate des erreurs d'étiquetage après approbation ? Les erreurs découvertes après l'approbation doivent être signalées via le système MedWatch de la FDA. Une fois le problème signalé, l'équipe qualité étudie le problème, en identifie la cause première et applique un correctif dans le cadre du flux de travail établi. La documentation de la manière dont la correction a été appliquée permet de garder le dossier prêt à être inspecté.
- Pourquoi le Code national des médicaments (NDC) doit-il figurer sur les étiquettes ? Le National Drug Code (NDC) est l'identifiant unique du produit, qui permet de le relier à l'enregistrement auprès de la FDA et d'éviter toute confusion lors de la distribution. Sur la ligne d'impression, il fournit les détails nécessaires pour confirmer que chaque boîte et chaque emballage correspondent à la bonne version du médicament.
- Qui examine et approuve les étiquettes des produits pharmaceutiques ? Aux États-Unis, le Center for Drug Evaluation and Research (CDER) de la FDA est responsable de l'examen et de l'approbation des étiquettes. Pour les substances contrôlées, la Drug Enforcement Administration (DEA) impose des exigences supplémentaires en matière de placement et d'avertissement. Les conseils sont disponibles directement via les ressources d'étiquetage de la FDA sur les portails Web officiels de l'agence.
- Quelles sont les sanctions en cas de non-conformité ? Les sanctions peuvent inclure des observations de la FDA-483, des lettres d'avertissement, des rappels ou la suspension de la fabrication. Ces mesures entraînent également des coûts financiers importants, notamment l'exécution des rappels, la perte de produits, l'arrêt de la production et les dépenses de remédiation. Le coût le plus élevé survient souvent par la suite, les entreprises étant confrontées à une atteinte à leur réputation et à une responsabilité potentielle. Ils risquent également de perdre l'accès au marché. Le respect des exigences en matière d'étiquetage permet d'éviter ces conséquences et de protéger les patients.
- Quels outils permettent de garantir la conformité de l'étiquetage ? Les équipes s'appuient souvent sur un logiciel de contrôle préalable associé à des outils tels que les plug-ins Adobe Acrobat ou InDesign pour vérifier les fichiers PDF avant de les imprimer. Ces outils confirment que les polices, les images, les profils ICC et les espaces colorimétriques sont corrects. Ils génèrent également un rapport préliminaire contenant des informations détaillées sur chaque correction et stockent chaque version afin que les audits puissent être effectués rapidement et que les enregistrements restent clairs.
- Pouvez-vous donner un exemple d'erreurs courantes détectées par les logiciels ? Les problèmes courants incluent les conflits de transparence, les logos manquants ou en basse résolution et les substitutions de polices dans les flux de travail PDF. Détecter ces problèmes avant que les fichiers ne soient imprimés réduit le nombre de réimpressions et garantit une qualité constante dans toutes les catégories de produits.
- Quelle documentation les entreprises doivent-elles conserver ? Les entreprises doivent conserver des archives contenant les étiquettes approuvées ainsi que les dossiers de soumission et la correspondance réglementaire. Les documents doivent être prêts à être inspectés à tout moment. De nombreuses organisations proposent également des cours internes afin que le personnel suive des pratiques cohérentes en matière de documentation.
- Qui est responsable de l'examen et de l'approbation des étiquettes pharmaceutiques en interne ? Alors que la FDA ou d'autres régulateurs accordent l'approbation finale, le processus d'examen interne est d'abord géré par les propres équipes de l'entreprise. Le personnel des affaires réglementaires et de l'assurance qualité dirige généralement et fait appel à une expertise supplémentaire lorsque la précision médicale, le risque juridique ou la conception de l'emballage nécessitent un examen plus approfondi. Chaque étape est documentée de sorte qu'au moment où une étiquette est soumise à la FDA, l'entreprise puisse montrer que chaque point de contrôle interne a été effectué.
Principales ressources :
- » de la FDAConsidérations de sécurité pour les étiquettes des contenants et la conception des étiquettes des cartons afin de minimiser les erreurs de médication« (mai 2022)
- «Étiquetage des médicaments sur ordonnance pour usage humain et des produits biologiques — Mise en œuvre des exigences de contenu et de format du PLR«
- FDA Division de la prévention et de l'analyse des erreurs médicamenteuses (pour des questions de conformité spécifiques)
Orientations futures de l'étiquetage des produits pharmaceutiques
La plupart des établissements cliniques ne s'appuient plus sur des informations de prescription sur papier. Au lieu de cela, distribution électronique fournit des données structurées sur l'étiquetage des produits que les systèmes pharmaceutiques utilisent pour générer des alertes d'interaction médicamenteuse et étayer les décisions cliniques. Les codes QR relient l'emballage physique au contenu numérique qui ne tient pas sur les contenants. Il ne s'agit pas de technologies émergentes, mais de la réalité du fonctionnement actuel de l'étiquetage.
Harmonisation mondiale grâce à l'ICH, les exigences de base en matière de documentation ont été harmonisées, mais les différences régionales restent bien ancrées. Bien que des efforts tels que le format XML PM du Canada se rapprochant du SPL de la FDA montrent des progrès, les différences entre les exigences relatives aux brochures destinées aux patients et les directives relatives aux produits biologiques restent importantes d'une juridiction à l'autre. L'EMA exige des brochures d'information destinées aux patients dans toutes les langues officielles de l'UE, avec des adaptations spécifiques au marché. Le PMDA japonais impose des exigences distinctes en matière de notice d'emballage. Les entreprises qui gèrent des produits sur plusieurs marchés gèrent toujours plusieurs versions d'étiquettes pour des médicaments identiques, bien que de meilleurs systèmes de données aient réduit les erreurs créées par le suivi manuel.
Accessibilité des patients est un domaine en perpétuel changement. Les régulateurs vont au-delà des déclarations générales concernant le langage clair et définissent désormais plus clairement ce qui constitue une communication adaptée aux besoins des patients, plutôt que ce qui répond simplement à une case de conformité. Les formats numériques ouvrent la voie à la personnalisation. Un patient peut ajuster la taille de la police pour plus de lisibilité, passer à sa langue préférée ou choisir une version audio. Ces outils ne sont pas encore officiellement reconnus à des fins réglementaires. Les autorités américaines et canadiennes procèdent actuellement à des examens législatifs et réglementaires, ce qui signifie que l'étiquetage adaptatif demeure une norme évolutive plutôt qu'une pratique établie.
Dans le même temps, les régulateurs s'appuient de plus en plus sur preuves du monde réel. Les données provenant des dossiers médicaux électroniques et des registres de patients jouent désormais un rôle croissant dans l'élaboration des exigences en matière d'étiquetage et des décisions d'approbation. Au fur et à mesure que les agences intègrent des données réelles à la surveillance de la sécurité, les mises à jour des étiquettes peuvent se faire plus rapidement que le cycle annuel traditionnel. Le véritable test sera la rapidité avec laquelle les fabricants adapteront leurs systèmes, car les changements fréquents augmentent le risque de défaillances du contrôle de version et d'erreurs d'étiquetage.
La solution Verify de GlobalVision est conçue pour relever les défis d'inspection auxquels sont confrontées les équipes pharmaceutiques lorsque la conformité ne peut pas attendre qu'une erreur humaine apparaisse. Découvrez comment fonctionne Verify.
