
A mislabeled vial slips through every checkpoint and reaches a hospital pharmacy. The dosage instructions look routine - a single digit off that no one notices until it’s too late. It’s a stark reminder that compliant pharmaceutical labeling remains one of the most critical safeguards in the pharmaceutical industry.
Introduction to Pharmaceutical Labeling Compliance
Compliant pharmaceutical labeling serves as the direct line between pharmaceutical companies and healthcare professionals, delivering essential information that directly impacts patient safety. This includes everything visible on drug packaging and containers along with accompanying materials, ensuring medical professionals and patients have accurate information to make safe, informed treatment decisions.
Regulatory bodies worldwide maintain strict guidelines because labeling errors can cause serious harm. In the United States, the Food and Drug Administration (FDA) defines and enforces pharmaceutical labeling requirements for both prescription drugs and medical devices. Across the European Union, this role falls to the European Medicines Agency (EMA). In coordination with Health Canada, Japan’s Pharmaceuticals and Medical Devices Agency (PDMA), and Therapeutic Goods Administration (TGA), these regulatory agencies create frameworks designed to protect public health and uphold consistency across markets. Efforts to harmonize standards through the International Council for Harmonisation (ICH) continue, but full alignment remains out of reach, with each region applying its own regulatory requirements that pharmaceutical companies must manage carefully. Meanwhile, the regulatory landscape keeps evolving: digital information delivery is changing how healthcare providers access data, and serialization mandates are redefining how pharmaceutical labeling processes function across the industry.
Key Regulatory Frameworks for Pharmaceutical Labeling
Since 2006, the FDA’s Physician Labeling Rule (PLR) has defined the modern standard for prescription drug labeling, bringing consistency and structure to every approved product. It introduced a clear hierarchy for prescribing information, featuring a concise “Highlights” section for healthcare practitioners and specific rules for how the remaining content must appear. These regulations respond directly to medication errors that occur when critical information gets buried in dense text. These strict regulations apply across product categories, including over the counter medications that consumers access without prescriptions.
International standards add layers of requirements for companies operating across markets. The EU's Summary of Product Characteristics (SmPC) and Package Leaflet requirements differ from FDA formats in both structure and content emphasis. While ICH guidelines aim to harmonize technical requirements, each region maintains distinct approaches. The table below shows how major markets handle key labeling requirements differently.
Regulatory labeling requirements across major markets

Japan mandates that all labeling content be written in Japanese and formatted to match local medical documentation practices. Australia’s TGA defines its own requirements for the Australian Register of Therapeutic Goods, reflecting national standards for clarity and consistency. Companies launching pharmaceutical products globally must maintain multiple label versions, each meeting local regulatory standards while ensuring consistency in core safety messaging. Managing version control across these markets requires systems that track which label version applies where, making pharma labeling compliance a discipline that combines regulatory affairs expertise with operational precisions to navigate today’s global regulatory maze.
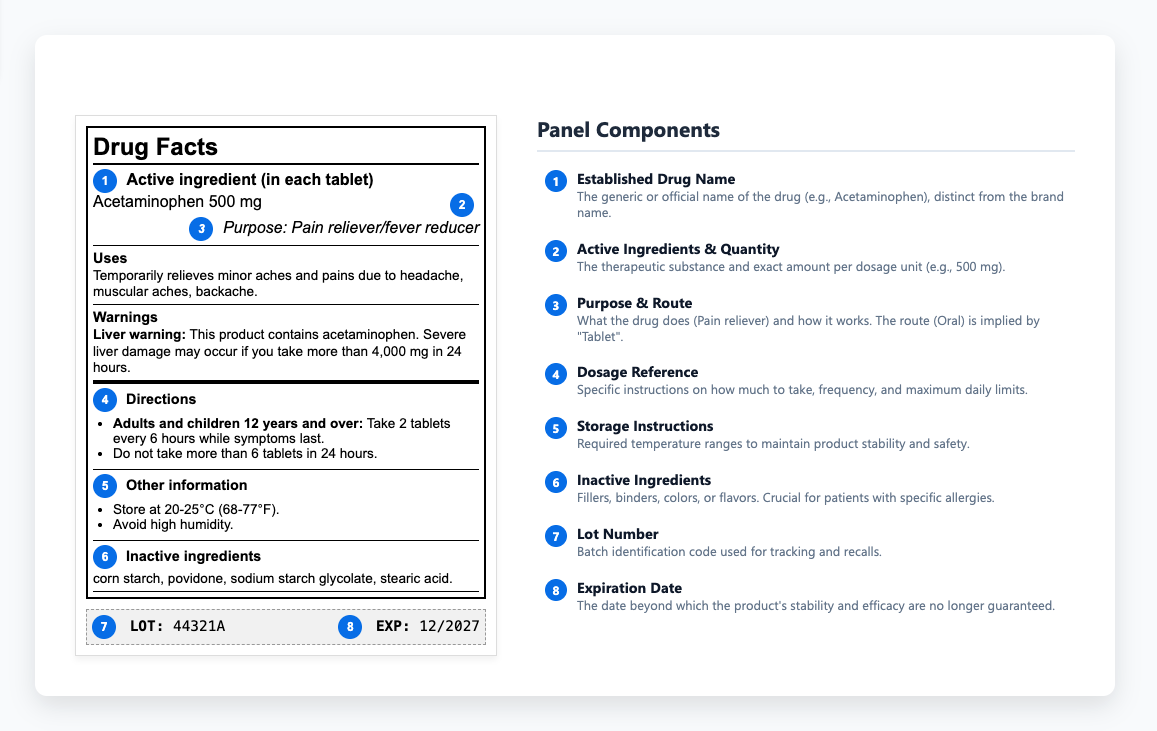
Essential Components of Compliant Prescription Drug Labels
Prescription drug labels deliver different information depending on where they appear. The immediate container label - what's on the bottle or vial - covers the basics: drug name, dosage form, strength, route of administration, and lot number. Carton labeling provides more room to work with. The principal display panel shows the established name and strength along with package quantity. Side panels handle prescribing information highlights and patient counseling details, plus manufacturer information. Regulations specify where everything appears and how prominently, down to exact font sizes that make product labeling an exacting discipline.
It starts with highlights that surface critical prescribing and safety information, then moves through the complete prescribing information. Each section follows a prescribed format, and any deviation requires justification and regulatory approval. The structure keeps essential safety data consistent and easy to find, helping reviewers and healthcare practitioners access the right details quickly. Key sections include:
- Indications and usage
- Dosage and administration
- Contraindications
- Warnings and precautions
- Adverse reactions
- Drug interactions
- Use in specific populations
- Clinical pharmacology
Patient information sections need to hit a sixth to eighth-grade reading level in the US, as outlined in FDA readability guidelines. Other regulatory agencies may apply different literacy or formatting standards, reflecting language and healthcare context in their regions. Medication guides for drugs with serious risks help patients understand safe use without overwhelming them with technical details. Clear usage instructions and dosage instructions show patients how to take their medications correctly and what to watch for.
Understanding the Drug Information Panel
The drug information panel provides healthcare practitioners with the details they need to prescribe and administer medications safely. Within that content, storage instructions carry special weight because they determine whether a drug maintains its stability and effectiveness throughout its shelf life. To protect that stability, labels define required temperature ranges and explain how to shield products from light and humidity during storage or transport. When those limits are overlooked, degradation can occur before a product ever reaches patients - a preventable loss of quality that underscores the need for thorough QA review. Some medications must be refrigerated, others protected from light, and many lose potency if exposed to moisture in transit. These requirements determine whether products stay effective or turn into useless (or harmful) substances. When these instructions get communicated unclearly or ignored entirely, products lose efficacy or become potentially harmful.
When formulations change or new safety data appears, label updates sometimes lag behind, creating mismatches between approved submissions and the labels released to market. Those inconsistencies may seem minor at first, but they often surface during regulatory inspections, slowing compliance reviews and adding unnecessary risk. Auditors compare label content directly against the approved versions, so these issues rarely stay hidden for long.
Warning Labels and Safety Information
Black box warnings are the FDA’s most serious form of safety alert, used to emphasize life-threatening or severe risks that must appear prominently on prescription drug labels. They show up when standard precautions fail to prevent serious harm. The FDA adds them based on post-market data revealing patterns of dangerous adverse reactions, sometimes including deaths. These warnings sit at the top of prescribing information in bold borders, using direct language about what the risk is and who's most vulnerable. Contraindications go a step further by identifying when the drug absolutely shouldn't be used. This includes dangerous interactions with other medications, documented pregnancy risks, and pre-existing medical conditions that would make the drug unsafe.
Adverse-reaction information spans mild, expected effects through to rare but serious complications. Presenting frequency data alongside those outcomes helps put the level of risk into context for patients and reviewers. To reduce that risk, mitigation strategies often include adjusting doses for specific populations, applying monitoring protocols, and guiding health-care providers on appropriate counseling and follow-up. For some high-risk drugs, Risk Evaluation and Mitigation Strategies (REMS) programs mandate additional labeling elements to ensure patient safety as binding requirements.

Specialized Labeling Requirements for Generic Drugs
Generic drug labeling has to match the branded product's label almost exactly, a requirement the FDA enforces to ensure generics deliver the same safety and efficacy as the original. You can differ on a few things: patent information gets left out since it belongs to the brand, while your own manufacturer details and NDC codes replace theirs. The "How Supplied" section can also reflect your specific packaging.
The real work starts when the branded label changes. A new warning or contraindication gets added based on post-market data, and your generic label needs to match. The FDA's "Changes Being Effected" pathway lets you make certain safety updates without waiting for approval, but you still own the responsibility to catch these changes and act on them. Companies that rely on regulators to tell them when labels are outdated end up behind, fixing problems after regulatory inspections flag them rather than staying ahead. Staying current means tracking when reference products change, then assessing how those updates affect your specific products. Updates need to roll out across manufacturing sites with documentation that proves compliance during audits. The pressure builds when safety updates need immediate action but your normal processes take months. Generic manufacturers who handle this well build monitoring systems that catch changes early rather than waiting for problems to surface during labeling compliance reviews.
Biological Products: Specific Labeling Considerations
Biological products come from living organisms instead of chemical synthesis, which changes what needs to appear on labels. Biologics require details about their biological source and manufacturing processes along with warnings about immune system responses that chemical drugs don't trigger.
Key Labeling Differences
Storage requirements for biologics go beyond standard specifications because these products degrade easily under conditions that wouldn't affect chemical drugs. Most need refrigeration within tight temperature ranges, and brief exposure to heat, light, or agitation can make them ineffective or potentially harmful. Labels outline how products should be stored and transported, detailing temperature limits and handling steps confirmed during development.
Immunogenicity warnings explain how the immune system can react to biological products. Labels explain the risk of developing anti-drug antibodies and summarize how clinical trials assessed that risk, including what the findings mean for patients. Healthcare providers use this information to monitor treatment response and adjust therapy when immune reactions develop. Post-market surveillance extends beyond standard adverse event reporting, with labels specifying registry participation requirements or long-term monitoring that tracks effects clinical trials couldn't fully capture.
Multilingual Labeling Challenges and Solutions
Pharmaceutical companies operating globally face distinct challenges when creating labels that meet diverse language requirements while maintaining accuracy and regulatory compliance.
Challenge: Translation quality and cultural accuracy
Medical terminology doesn't translate directly across languages, and cultural context shapes how patients understand health information. A technically accurate translation can completely miss how people in a target market actually discuss medical concepts. Standard translation services lack the specialized knowledge pharmaceutical labeling demands.
Effective translation requires specialists who understand both medical terminology and the regulatory expectations of each target market. Translators need native fluency and working knowledge of the pharmaceutical domain to ensure accuracy and context. The FDA and other regulatory agencies outline when translated materials need formal approval or a certified translator, with specific rules varying by region.
Challenge: Meeting diverse market language requirements
Many markets mandate multiple languages on a single label. Canada requires bilingual English and French. Switzerland needs German, French, and Italian. Belgium requires French, Dutch, and German. Fitting multiple complete translations onto space-constrained labels forces difficult tradeoffs between readability and meeting regulatory requirements. Companies address this by working with label designers who specialize in multilingual pharmaceutical packaging, using techniques like accordion folds, booklet labels, or tiered information hierarchies that prioritize critical safety information.
Challenge: Coordinating updates across languages and markets
When safety information changes on the master label, every translated version needs updating. Regulatory timelines across markets rarely align, creating periods where different countries show different safety information on their labels.
Technology provides solutions here. Translation memory systems maintain terminology consistency across products while terminology databases ensure technical terms translate uniformly every time they appear. Workflow management systems bring this together by tracking which label versions exist in which languages, which markets have approved each version, and where updates are needed. These systems help companies maintain compliance by flagging gaps and coordinating version control across global operations before regulatory inspections surface problems.
Serialization and Track & Trace Requirements
Serialization regulations changed how pharmaceutical labeling works by requiring every package to carry a unique identifier that tracks it through the supply chain. This addresses real problems, including counterfeiting and the need to execute rapid recalls when safety issues emerge. Regulations worldwide now mandate serialization, though implementation timelines have varied by market. The US Drug Supply Chain Security Act required compliance by 2023, while the EU's Falsified Medicines Directive took effect in 2019. Other markets followed their own schedules, creating a staggered global rollout that required companies to manage different requirements simultaneously. These regulations require packages to carry product identifiers with multiple data elements - National Drug Codes and unique serial numbers along with lot numbers and expiration dates - all in formats both humans and machines can read.
The technology depends mainly on 2D Data Matrix barcodes, which store large amounts of information in a compact format. These codes appear across every stage of production, from manufacturing lines and quality control systems to distribution databases that trace each unique identifier. For most teams, the real complexity comes when these requirements must coexist with labels already packed with mandatory content. Barcodes can’t obscure required text, and serial numbers must appear in both human- and machine-readable formats. At the same time, serialization data needs to remain in its traditional positions to comply with long-standing pharmaceutical labeling processes. In practice, companies are now designing labels that satisfy two generations of regulatory expectations while maintaining clarity and readability.
Quality Control in Pharmaceutical Labeling
Label design reviews compare proposed labels against approved regulatory submissions. Text needs to match exactly, graphics must appear as approved, formatting has to follow specifications. Even minor deviations require documentation and often trigger regulatory notification, which is why these reviews establish the baseline for all subsequent verification. Once vendor proofs arrive, AI-powered inspection tools help incoming quality assurance teams verify that files match approved master documents before printing begins:
- Artwork comparison checks proofs against approved masters to catch layout shifts or missing elements
- Barcode and QR code verification confirms that encoded data is correct and that each code follows required formatting before printing
- Optical character recognition (OCR) identifies text differences between proofs and approved master files to ensure no unintended changes were introduced during updates
- Intended change detection checks whether annotated corrections from earlier file versions were applied correctly, helping teams confirm that requested updates were implemented before production.
These systems work continuously during production runs, analyzing artwork and text to flag problems immediately rather than discovering them after thousands of units have been printed. Human oversight handles exceptions and provides final approval, but automated inspection carries the bulk of verification work at scale.
Documentation ties everything together by proving to regulators that procedures were actually followed. Batch records capture what happens during production - which inspections ran, what results came back, how deviations were resolved, who approved release. During regulatory inspections, these records demonstrate that your quality control system actually functions rather than just existing as written procedures. Missing documentation creates compliance concerns even when labels themselves are correct, because regulators can't verify the process worked as designed. Companies that integrate these quality control measures from design through documentation catch labeling errors before they become market problems, while those relying on reactive checks discover issues during audits when fixes are far more expensive.
Automation Tools for Compliant Labeling
These platforms manage workflows where multiple teams review labels before production, ensuring compliant labeling across every stage of review. Content moves from research and product development through regulatory review, then into design and marketing stages for layout, branding and claims verification before reaching quality assurance for final accuracy checks. The system tracks who reviewed what and when, creating the audit trail regulatory inspections demand. Companies can scale these workflows beyond what email chains and shared drives handle.
Validation follows the same rigor required for any pharmaceutical manufacturing system. The process starts with installation qualification to confirm correct setup, moves through operational qualification that tests all functions, and concludes with performance qualification demonstrating consistent accuracy during normal operations. Validation proves the automated system reliably prevents labeling errors rather than introducing new ones.
Integration with regulatory information management systems connects approved content directly to label production. Updates to prescribing information after FDA approvals flow straight to relevant labels without anyone manually copying text between systems, which cuts transcription errors while accelerating change cycles. The main benefits include faster label updates and tighter version control across global sites, supported by structured review workflows that strengthen overall regulatory compliance. Companies see operational efficiency gains by reducing manual touchpoints and getting real-time visibility into labeling status across manufacturing operations.
Best Practices for Regulatory Submissions
Involve regulatory affairs during product development so labeling considerations can shape how you design clinical trials and collect data. Waiting until filing time means you're stuck trying to make your data support labeling claims it wasn't designed to prove.
Your submission needs labeling in the format the agency expects. The FDA wants structured product labeling (SPL) format, an XML-based standard their systems can process electronically. Content follows industry standards - sections appear in prescribed order with all required elements included. Get the technical specs wrong and your submission gets rejected over formatting rather than any actual safety or efficacy concerns.
Tracking what changed from your currently approved version separates smooth labeling updates from regulatory headaches. Some changes need prior approval before implementation, others go through under "changes being effected" provisions, and some just get reported annually. Mix these up and you're creating compliance problems that slow down getting updates to market. Electronic submissions have replaced paper, with the FDA's Gateway processing structured content differently than PDFs. Many companies bring in specialized services to handle the technical requirements and ensure accuracy. After approval, your systems need to guarantee only approved versions reach production. Change control that actually works prevents old labels from slipping through after new versions get approved.
Training and Documentation Requirements
Everyone involved in labeling operations needs to understand what they're doing and why it matters, and you need to prove it. These sessions aren’t one-time events. Continuous training keeps teams current with regulatory changes and new systems. The real measure isn’t whether training happens, but whether teams understand the current requirements when inspectors arrive.
Standard operating procedures (SOPs) document how your company handles every step of the labeling process, showing regulators you follow a defined process rather than making decisions differently depending on who's working that day. SOPs need to cover:
- How artwork files get managed and version controlled
- How approvals flow through the organization
- How changes get initiated, reviewed, and implemented
- How deviations and problems get investigated and resolved
Writing SOPs is straightforward; following them consistently and updating them when labeling practices change is where companies struggle. Records demonstrate that your quality system functions as designed, not just on paper. They need to show that trained personnel followed approved procedures, inspections were completed as planned, and any deviations were properly investigated. During regulatory inspections, these files answer the most basic question: did you actually do what your procedures say you do? Training extends beyond labeling specialists to anyone touching the process, from regulatory affairs staff writing content to graphic designers creating artwork to quality assurance personnel approving production runs. Teams that emphasize strong training programs tend to have fewer compliance issues during audits and can respond faster when requirements change. The alternative is discovering gaps when regulators find them, which costs far more to fix.
Future Trends in Pharmaceutical Labeling
Digital labeling is moving beyond PDF versions of paper inserts. QR codes on packages now link directly to prescribing information that updates in real time, letting healthcare professionals access current safety data rather than relying on printed inserts that might be months old. The FDA recognizes paper labels have limits, particularly for complex products where comprehensive information exceeds reasonable space constraints, but regulators haven't abandoned physical labeling. Core safety information still requires presence on packaging to ensure access without digital devices.
Environmental pressure is reshaping packaging decisions in ways that sometimes conflict with regulatory requirements. The pharmaceutical sector faces demands to reduce waste and use sustainable materials, goals that can clash with regulations specifying particular packaging constructions. The European Union has pushed particularly aggressive sustainable packaging requirements that affect label design and production.
Smart labels with NFC chips or embedded technologies verify product authenticity while tracking usage and monitoring temperature exposure throughout distribution. These capabilities raise new regulatory questions about data privacy and security, as well as how far smart label functionality extends into medical device territory. The technology exists; the regulatory framework for it is still forming.
Resources for Pharmaceutical Labeling Professionals
Professional associations like the Regulatory Affairs Professionals Society (RAPS) and the Drug Information Association (DIA) connect you with people facing similar labeling challenges through conferences and working groups. You get insight into how regulators approach new requirements while seeing how other companies solve problems you're dealing with. RAPS also offers regulatory affairs certification that covers labeling competencies, while specialized training programs address specific gaps when teams need to get current on new regulations or technologies.
External support comes from two directions. Consulting firms bring regulatory expertise for navigating complex submissions, particularly when you're dealing with unfamiliar requirements or tight deadlines. Technology providers offer platforms that handle version management across markets while coordinating changes and approvals. The choice between them often depends on whether your challenge is understanding what regulators want or managing the operational complexity of getting it done.
For teams managing high-stakes content, precision isn’t optional. GlobalVison’s Verify gives teams the confidence that every approved version is inspection-ready.
