
Ein falsch etikettiertes Fläschchen rutscht durch jeden Checkpoint und erreicht eine Krankenhausapotheke. Die Dosierungsanweisungen sehen routinemäßig aus — eine einstellige Ziffer, die niemand bemerkt, bis es zu spät ist. Es ist eine deutliche Erinnerung daran, dass die gesetzeskonforme Arzneimittelkennzeichnung nach wie vor eine der wichtigsten Schutzmaßnahmen in der Pharmaindustrie ist.
Einführung in die Einhaltung der Vorschriften zur Arzneimittelkennzeichnung
Die konforme Arzneimittelkennzeichnung dient als direkte Verbindung zwischen Pharmaunternehmen und medizinischem Fachpersonal und liefert wichtige Informationen, die sich direkt auf die Patientensicherheit auswirken. Dazu gehört alles, was auf Arzneimittelverpackungen und -behältern sowie auf den Begleitmaterialien sichtbar ist. So wird sichergestellt, dass medizinisches Fachpersonal und Patienten über genaue Informationen verfügen, um sichere und fundierte Behandlungsentscheidungen treffen zu können.
Aufsichtsbehörden weltweit halten strenge Richtlinien ein, da Kennzeichnungsfehler schwerwiegende Schäden verursachen können. In den Vereinigten Staaten Lebensmittel- und Arzneimittelbehörde (FDA) definiert und durchsetzt pharmazeutische Kennzeichnungsanforderungen sowohl für verschreibungspflichtige Medikamente als auch für Medizinprodukte. In der gesamten Europäischen Union fällt diese Rolle dem zu Europäische Arzneimittel-Agentur (EMMA). In Abstimmung mit Gesundheit Kanada, Japans Behörde für Arzneimittel und Medizinprodukte (PDMA) und Verwaltung therapeutischer Güter (TGA) schaffen diese Aufsichtsbehörden Rahmenbedingungen zum Schutz der öffentlichen Gesundheit und zur Wahrung der Marktkonsistenz. Bemühungen zur Harmonisierung der Standards durch die Internationaler Rat für Harmonisierung (ICH) fortsetzen, aber eine vollständige Angleichung ist nach wie vor unerreichbar, da jede Region ihre eigenen regulatorischen Anforderungen anwendet, die Pharmaunternehmen sorgfältig verwalten müssen. In der Zwischenzeit entwickelt sich das regulatorische Umfeld ständig weiter: Die digitale Bereitstellung von Informationen verändert die Art und Weise, wie Gesundheitsdienstleister auf Daten zugreifen, und Serialisierungsvorschriften definieren die Funktionsweise pharmazeutischer Kennzeichnungsprozesse in der gesamten Branche neu.
Wichtige regulatorische Rahmenbedingungen für die Arzneimittelkennzeichnung
Seit 2006 ist die FDA Kennzeichnungsregel für Ärzte (PLR) hat den modernen Standard für definiert rezeptpflichtiges Medikament Etikettierung, die jedem zugelassenen Produkt Konsistenz und Struktur verleiht. Es wurde eine klare Hierarchie für Verschreibungsinformationen eingeführt, einschließlich eines kurzen Abschnitts mit „Highlights“ für Ärzte und speziellen Regeln für das Erscheinungsbild der übrigen Inhalte. Diese Vorschriften reagieren direkt auf Medikationsfehler, die auftreten, wenn wichtige Informationen in dichtem Text untergehen. Diese strengen Vorschriften gelten für alle Produktkategorien, einschließlich rezeptfreier Medikamente, zu denen Verbraucher ohne Rezept greifen.
Internationale Standards fügen zusätzliche Anforderungen für Unternehmen hinzu, die marktübergreifend tätig sind. Die der EU Zusammenfassung der Produktmerkmale (SmPC) und die Packungsbeilagenanforderungen unterscheiden sich sowohl in der Struktur als auch in der inhaltlichen Ausrichtung von den FDA-Formaten. Die ICH-Richtlinien zielen zwar darauf ab, die technischen Anforderungen zu harmonisieren, doch in jeder Region werden unterschiedliche Konzepte verfolgt. Die folgende Tabelle zeigt, wie die wichtigsten Märkte die wichtigsten Kennzeichnungsanforderungen unterschiedlich behandeln.
Regulatorische Kennzeichnungsanforderungen in allen wichtigen Märkten

Japan schreibt vor, dass alle Kennzeichnungsinhalte auf Japanisch verfasst und so formatiert sind, dass sie den örtlichen medizinischen Dokumentationspraktiken entsprechen. Die australische TGA definiert ihre eigenen Anforderungen an das Australian Register of Therapeutic Goods, die den nationalen Standards für Klarheit und Einheitlichkeit entsprechen. Unternehmen, die weltweit pharmazeutische Produkte auf den Markt bringen, müssen mehrere Versionen auf dem Etikett verwenden, von denen jede den lokalen regulatorischen Standards entspricht und gleichzeitig die Konsistenz der wichtigsten Sicherheitsinformationen gewährleistet. Verwaltung Versionskontrolle In diesen Märkten sind Systeme erforderlich, die verfolgen, welche Etikettenversion wo gilt, was die Einhaltung der Arzneimittelkennzeichnung zu einer Disziplin macht, die Fachwissen in regulatorischen Angelegenheiten mit operativer Präzision kombiniert, um sich im heutigen globalen regulatorischen Labyrinth zurechtzufinden.
Wesentliche Bestandteile gesetzeskonformer Etiketten für verschreibungspflichtige Medikamente
Etiketten für verschreibungspflichtige Medikamente enthalten unterschiedliche Informationen, je nachdem, wo sie erscheinen. Das Etikett auf der Verpackung — also das, was auf der Flasche oder dem Fläschchen steht — deckt die grundlegenden Informationen ab: Name des Arzneimittels, Darreichungsform, Stärke, Verabreichungsweg und Chargennummer. Die Etikettierung auf dem Karton bietet mehr Platz zum Arbeiten. Auf dem Hauptanzeigefeld werden der festgelegte Name und die Stärke sowie die Verpackungsmenge angezeigt. Auf den Seitenwänden befinden sich die wichtigsten Verschreibungsinformationen und Informationen zur Patientenberatung sowie Herstellerinformationen. Die Vorschriften legen fest, wo und wie deutlich alles erscheint, bis hin zu den genauen Schriftgrößen, die die Produktkennzeichnung zu einer anspruchsvollen Disziplin machen.
Es beginnt mit Markierungen, die wichtige Verschreibungs- und Sicherheitsinformationen enthalten, und geht dann durch die vollständigen Verschreibungsinformationen. Jeder Abschnitt folgt einem vorgeschriebenen Format, und jede Abweichung bedarf einer Begründung und behördlichen Genehmigung. Die Struktur sorgt dafür, dass wichtige Sicherheitsdaten konsistent und leicht auffindbar sind, sodass Prüfer und medizinisches Fachpersonal schnell auf die richtigen Informationen zugreifen können. Zu den wichtigsten Abschnitten gehören:
- Indikationen und Verwendung
- Dosierung und Verabreichung
- Gegenanzeigen
- Warnungen und Vorsichtsmaßnahmen
- Unerwünschte Reaktionen
- Arzneimittelwechselwirkungen
- Verwendung in bestimmten Populationen
- Klinische Pharmakologie
Die Abschnitte mit Patienteninformationen müssen in den USA ein Leseniveau der sechsten bis achten Klasse erreichen, wie in den Lesbarkeitsrichtlinien der FDA beschrieben. Andere Aufsichtsbehörden wenden möglicherweise andere Lese-, Schreib- oder Formatierungsstandards an, die der Sprache und dem medizinischen Kontext in ihren Regionen entsprechen. Leitfäden für Medikamente mit schwerwiegenden Risiken helfen Patienten dabei, ihre sichere Anwendung zu verstehen, ohne sie mit technischen Details zu überfordern. Klare Anwendungs- und Dosierungsanweisungen zeigen den Patienten, wie sie ihre Medikamente richtig einnehmen und worauf sie achten müssen.
Das Arzneimittelinformationsgremium verstehen
Das Arzneimittelinformationspanel bietet Ärzten die Informationen, die sie benötigen, um Medikamente sicher zu verschreiben und zu verabreichen. Innerhalb dieses Inhaltes haben die Hinweise zur Aufbewahrung ein besonderes Gewicht, da sie bestimmen, ob ein Medikament seine Stabilität und Wirksamkeit während seiner gesamten Haltbarkeitsdauer beibehält. Um diese Stabilität zu gewährleisten, definieren die Etiketten die erforderlichen Temperaturbereiche und erklären, wie Produkte während der Lagerung oder des Transports vor Licht und Feuchtigkeit geschützt werden können. Werden diese Grenzwerte nicht eingehalten, kann es zu einer Verschlechterung kommen, noch bevor ein Produkt die Patienten erreicht — ein vermeidbarer Qualitätsverlust, der die Notwendigkeit einer gründlichen Qualitätsprüfung unterstreicht. Einige Medikamente müssen gekühlt, andere vor Licht geschützt werden, und viele verlieren an Wirksamkeit, wenn sie während des Transports Feuchtigkeit ausgesetzt werden. Diese Anforderungen bestimmen, ob Produkte wirksam bleiben oder sich in nutzlose (oder schädliche) Substanzen verwandeln. Wenn diese Anweisungen unklar kommuniziert oder gänzlich ignoriert werden, verlieren Produkte ihre Wirksamkeit oder werden potenziell schädlich.
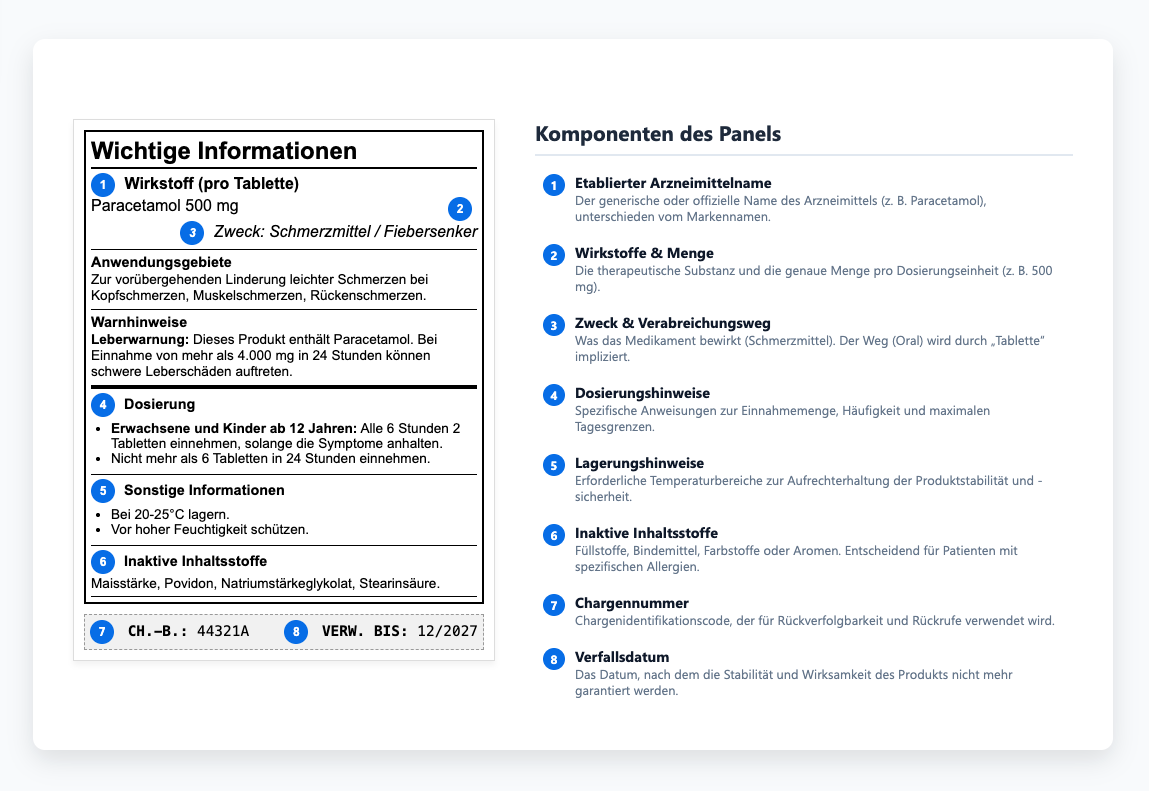
Wenn sich Rezepturen ändern oder neue Sicherheitsdaten erscheinen, hinken die Aktualisierungen der Etiketten manchmal hinterher, was zu Diskrepanzen zwischen den zugelassenen Einreichungen und den auf den Markt gebrachten Etiketten führt. Diese Inkonsistenzen mögen auf den ersten Blick geringfügig erscheinen, treten aber häufig bei behördlichen Inspektionen zutage, was die Überprüfung der Einhaltung der Vorschriften verlangsamt und unnötige Risiken mit sich bringt. Auditoren vergleichen den Inhalt der Etiketten direkt mit den genehmigten Versionen, sodass diese Probleme selten lange verborgen bleiben.
Warnetiketten und Sicherheitsinformationen
Blackbox-Warnungen sind die schwerwiegendste Form der Sicherheitswarnung der FDA. Sie werden verwendet, um auf lebensbedrohliche oder schwerwiegende Risiken hinzuweisen, die auf den Etiketten verschreibungspflichtiger Medikamente deutlich sichtbar erscheinen müssen. Sie tauchen auf, wenn die üblichen Vorsichtsmaßnahmen schwere Schäden nicht verhindern. Die FDA fügt sie auf der Grundlage von Daten nach Markteinführung hinzu, die Muster gefährlicher Nebenwirkungen, manchmal auch Todesfälle, aufdecken. Diese Warnhinweise stehen in fett umrandeten Umrandungen am Anfang der Verschreibungsinformationen und geben direkt Auskunft darüber, um welches Risiko es sich handelt und wer am stärksten gefährdet ist. Gegenanzeigen gehen noch einen Schritt weiter und geben an, wann das Medikament auf keinen Fall eingenommen werden sollte. Dazu gehören gefährliche Wechselwirkungen mit anderen Medikamenten, dokumentierte Schwangerschaftsrisiken und bereits bestehende Erkrankungen, die das Medikament unsicher machen würden.
Die Informationen zu Nebenwirkungen reichen von leichten, erwarteten Wirkungen bis hin zu seltenen, aber schwerwiegenden Komplikationen. Die Präsentation von Häufigkeitsdaten zusammen mit diesen Ergebnissen hilft Patienten und Gutachtern dabei, das Risikoniveau in einen Kontext zu stellen. Um dieses Risiko zu verringern, umfassen die Strategien zur Risikominderung häufig die Anpassung der Dosen für bestimmte Bevölkerungsgruppen, die Anwendung von Überwachungsprotokollen und die Anleitung von Gesundheitsdienstleistern zu angemessenen Beratungs- und Nachsorgemaßnahmen. Bei einigen Medikamenten mit hohem Risiko gilt Folgendes: Strategien zur Risikobewertung und Minderung (REMS) -Programme schreiben zusätzliche Kennzeichnungselemente zur Gewährleistung der Patientensicherheit als verbindliche Anforderungen vor.

Spezialisierte Kennzeichnungsanforderungen für Generika
Die Kennzeichnung von Generika muss fast exakt mit dem Etikett des Markenprodukts übereinstimmen, eine Anforderung, die die FDA durchsetzt, um sicherzustellen, dass Generika die gleiche Sicherheit und Wirksamkeit wie das Original bieten. In einigen Dingen können Sie unterschiedlicher Meinung sein: Patentinformationen werden weggelassen, da sie zur Marke gehören, während Ihre eigenen Herstellerangaben und NDC-Codes ihre eigenen ersetzen. Der Abschnitt „Wie geliefert“ kann sich auch auf Ihre spezielle Verpackung beziehen.
Die eigentliche Arbeit beginnt, wenn sich das Markenlabel ändert. Basierend auf Daten nach dem Inverkehrbringen wird ein neuer Warnhinweis oder eine neue Gegenanzeige hinzugefügt, und Ihr generisches Etikett muss dazu passen. Das FDA-Verfahren „Änderungen werden bewirkt“ ermöglicht es Ihnen, bestimmte Sicherheitsaktualisierungen vorzunehmen, ohne auf die Zulassung warten zu müssen. Sie sind jedoch weiterhin dafür verantwortlich, diese Änderungen zu erkennen und entsprechend zu handeln. Unternehmen, die sich darauf verlassen, dass die Aufsichtsbehörden ihnen sagen, wenn Kennzeichnungen veraltet sind, hinken hinterher und beheben Probleme, nachdem behördliche Inspektionen sie festgestellt haben, anstatt die Nase vorn zu haben. Um auf dem neuesten Stand zu bleiben, müssen Sie verfolgen, wann sich Referenzprodukte ändern, und dann beurteilen, wie sich diese Aktualisierungen auf Ihre spezifischen Produkte auswirken. Aktualisierungen müssen an allen Produktionsstandorten eingeführt werden, zusammen mit einer Dokumentation, die die Einhaltung der Vorschriften bei Audits belegt. Der Druck steigt, wenn Sicherheitsaktualisierungen sofortige Maßnahmen erfordern, Ihre normalen Prozesse jedoch Monate in Anspruch nehmen. Generikahersteller, die sich damit befassen, bauen gut Überwachungssysteme, die Änderungen frühzeitig erkennen, anstatt darauf zu warten, dass Probleme bei der Überprüfung der Einhaltung der Kennzeichnungsetiketten auftauchen.
Biologische Produkte: Spezifische Überlegungen zur Kennzeichnung
Biologische Produkte stammen aus lebenden Organismen und nicht aus chemischer Synthese, was die Angaben auf den Etiketten verändert. Biologika benötigen Informationen über ihre biologische Herkunft und Herstellungsverfahren sowie Warnungen vor Reaktionen des Immunsystems, die chemische Medikamente nicht auslösen.
Die wichtigsten Unterschiede bei der Kennzeichnung
Die Lagerungsanforderungen für Biologika gehen über die Standardspezifikationen hinaus, da sich diese Produkte unter Bedingungen, die chemische Medikamente nicht beeinträchtigen würden, leicht zersetzen. Die meisten müssen innerhalb enger Temperaturbereiche gekühlt werden, und kurze Einwirkung von Hitze, Licht oder Bewegung können dazu führen, dass sie unwirksam oder potenziell schädlich sind. Auf den Etiketten wird beschrieben, wie Produkte gelagert und transportiert werden sollten. Dabei sind die Temperaturgrenzwerte und die während der Entwicklung bestätigten Verarbeitungsschritte aufgeführt.
Immunogenitätswarnungen erklären, wie das Immunsystem auf biologische Produkte reagieren kann. Auf den Etiketten wird das Risiko der Entwicklung von Antikörpern gegen Medikamente erklärt und es wird zusammengefasst, wie dieses Risiko in klinischen Studien bewertet wurde, einschließlich der Bedeutung der Ergebnisse für die Patienten. Gesundheitsdienstleister verwenden diese Informationen, um das Ansprechen auf die Behandlung zu überwachen und die Therapie anzupassen, wenn sich Immunreaktionen entwickeln. Die Überwachung nach der Markteinführung geht über die übliche Meldung von Nebenwirkungen hinaus. Auf den Etiketten sind die Anforderungen an die Teilnahme an den Registern angegeben, oder es gibt eine Langzeitüberwachung, die Auswirkungen verfolgt, die in klinischen Studien nicht vollständig erfasst werden konnten.
Herausforderungen und Lösungen für die mehrsprachige Etikettierung
Pharmaunternehmen, die weltweit tätig sind, stehen vor besonderen Herausforderungen, wenn es darum geht, Etiketten zu erstellen, die unterschiedlichen Sprachanforderungen entsprechen und gleichzeitig die Genauigkeit und die Einhaltung gesetzlicher Vorschriften gewährleisten.
Herausforderung: Übersetzungsqualität und kulturelle Genauigkeit
Medizinische Terminologie lässt sich nicht direkt in mehrere Sprachen übersetzen, und der kulturelle Kontext bestimmt, wie Patienten Gesundheitsinformationen verstehen. Bei einer fachlich korrekten Übersetzung kann völlig übersehen werden, wie Menschen in einem Zielmarkt tatsächlich über medizinische Konzepte sprechen. Standardübersetzungsdiensten fehlt das Fachwissen, das für die Etikettierung von Arzneimitteln erforderlich ist.
Effektive Übersetzungen erfordern Spezialisten, die sowohl die medizinische Terminologie als auch die regulatorischen Erwartungen der einzelnen Zielmärkte verstehen. Übersetzer benötigen fließende Muttersprachkenntnisse und praktische Kenntnisse im pharmazeutischen Bereich, um Genauigkeit und Kontext zu gewährleisten. Die FDA und andere Aufsichtsbehörden legen fest, wann übersetzte Materialien eine formelle Genehmigung oder einen zertifizierten Übersetzer benötigen, wobei die spezifischen Regeln je nach Region variieren.
Herausforderung: Erfüllung der unterschiedlichen Anforderungen an die Marktsprache
Viele Märkte schreiben mehrere Sprachen auf einem einzigen Etikett vor. Kanada verlangt zweisprachiges Englisch und Französisch. Die Schweiz benötigt Deutsch, Französisch und Italienisch. Belgien benötigt Französisch, Niederländisch und Deutsch. Das Anbringen mehrerer vollständiger Übersetzungen auf Etiketten mit begrenztem Platzangebot führt zu schwierigen Kompromissen zwischen Lesbarkeit und Erfüllung behördlicher Anforderungen. Unternehmen begegnen diesem Problem, indem sie mit Etikettendesignern zusammenarbeiten, die sich auf mehrsprachige Arzneimittelverpackungen spezialisiert haben. Dabei verwenden sie Techniken wie Akkordeonfalten, Booklet-Etiketten oder abgestufte Informationshierarchien, bei denen wichtige Sicherheitsinformationen priorisiert werden.
Herausforderung: Sprach- und marktübergreifende Koordination von Updates
Wenn sich die Sicherheitsinformationen auf dem Master-Etikett ändern, muss jede übersetzte Version aktualisiert werden. Die regulatorischen Zeitpläne der Märkte stimmen selten überein, was zu Zeiträumen führt, in denen die verschiedenen Länder unterschiedliche Sicherheitsinformationen auf ihren Etiketten angeben.
Technologie bietet hier Lösungen. Translation-Memory-Systeme sorgen für eine produktübergreifende Terminologiekonsistenz, während Terminologiedatenbanken sicherstellen, dass Fachbegriffe bei jedem Erscheinen einheitlich übersetzt werden. Workflow-Management-Systeme führen dies zusammen, indem sie nachverfolgen, welche Labelversionen in welchen Sprachen existieren, welche Märkte die einzelnen Versionen freigegeben haben und wo Aktualisierungen erforderlich sind. Diese Systeme helfen Unternehmen dabei, die Einhaltung gesetzlicher Vorschriften aufrechtzuerhalten, indem sie Lücken aufzeigen und die Versionskontrolle im gesamten globalen Betrieb koordinieren, bevor behördliche Inspektionen Probleme auftauchen.
Anforderungen an Serialisierung und Track & Trace
Die Vorschriften zur Serialisierung haben die Funktionsweise der Arzneimittelkennzeichnung verändert, da jede Packung mit einer eindeutigen Kennzeichnung versehen sein muss, mit der sie durch die gesamte Lieferkette verfolgt werden kann. Dadurch werden echte Probleme behoben, darunter Fälschungen und die Notwendigkeit, bei Sicherheitsproblemen schnelle Rückrufe durchzuführen. Weltweit ist die Serialisierung inzwischen gesetzlich vorgeschrieben, obwohl die Umsetzungsfristen je nach Markt unterschiedlich sind. Der US-amerikanische Drug Supply Chain Security Act sah die Einhaltung bis 2023 vor, während die EU-Richtlinie über gefälschte Arzneimittel 2019 in Kraft trat. Andere Märkte folgten ihren eigenen Zeitplänen und führten zu einer gestaffelten globalen Einführung, bei der Unternehmen verschiedene Anforderungen gleichzeitig erfüllen mussten. Nach diesen Vorschriften müssen Verpackungen Produktidentifikatoren mit mehreren Datenelementen tragen — nationale Arzneimittelcodes und eindeutige Seriennummern sowie Chargennummern und Verfallsdaten — und das alles in Formaten, die sowohl Menschen als auch Maschinen lesen können.
Die Technologie basiert hauptsächlich auf 2D-Data-Matrix-Barcodes, die große Informationsmengen in einem kompakten Format speichern. Diese Codes kommen in jeder Phase der Produktion vor, von den Fertigungslinien und Qualitätskontrollsystemen bis hin zu Vertriebsdatenbanken, in denen jede eindeutige Kennzeichnung nachverfolgt werden kann. Für die meisten Teams entsteht die eigentliche Komplexität, wenn diese Anforderungen neben den Etiketten bestehen müssen, die bereits mit verbindlichen Inhalten ausgestattet sind. Barcodes können den erforderlichen Text nicht verdecken, und Seriennummern müssen sowohl in menschenlesbarer als auch in maschinenlesbarer Form erscheinen. Gleichzeitig müssen die Serialisierungsdaten an ihren traditionellen Positionen bleiben, um den seit langem bestehenden Verfahren zur Kennzeichnung pharmazeutischer Produkte zu entsprechen. In der Praxis entwerfen Unternehmen jetzt Etiketten, die den Anforderungen von zwei Generationen regulatorischer Vorschriften entsprechen und gleichzeitig Klarheit und Lesbarkeit gewährleisten.
Qualitätskontrolle bei der Arzneimittelkennzeichnung
Bei der Überprüfung des Etikettendesigns werden die vorgeschlagenen Etiketten mit den genehmigten behördlichen Einreichungen verglichen. Der Text muss exakt übereinstimmen, die Grafiken müssen als genehmigt erscheinen und die Formatierung muss den Spezifikationen entsprechen. Selbst geringfügige Abweichungen müssen dokumentiert werden und lösen häufig eine behördliche Meldung aus. Aus diesem Grund bilden diese Überprüfungen die Grundlage für alle nachfolgenden Überprüfungen. Sobald die Nachweise des Lieferanten eintreffen, helfen KI-gestützte Inspektionstools den Teams für die Qualitätssicherung, vor Druckbeginn zu überprüfen, ob die Dateien mit den genehmigten Masterdokumenten übereinstimmen:
- Vergleich von Kunstwerken vergleicht die Proofs mit zugelassenen Mastern, um Layoutverschiebungen oder fehlende Elemente zu erkennen
- Barcode- und QR-Code-Überprüfung bestätigt, dass die kodierten Daten korrekt sind und dass jeder Code vor dem Drucken der erforderlichen Formatierung folgt
- Optische Zeichenerkennung (OCR) identifiziert Textunterschiede zwischen Proofs und genehmigten Masterdateien, um sicherzustellen, dass bei Aktualisierungen keine unbeabsichtigten Änderungen vorgenommen wurden
- Erkennung beabsichtigter Änderungen prüft, ob die mit Anmerkungen versehenen Korrekturen aus früheren Dateiversionen korrekt angewendet wurden, sodass die Teams überprüfen können, ob die angeforderten Updates vor der Produktion implementiert wurden.
Diese Systeme arbeiten kontinuierlich während der Produktion und analysieren Grafiken und Text, um Probleme sofort zu erkennen, anstatt sie erst zu entdecken, nachdem Tausende von Einheiten gedruckt wurden. Die menschliche Aufsicht kümmert sich um Ausnahmen und erteilt die endgültige Genehmigung, aber die automatische Inspektion erledigt den Großteil der Verifizierungsarbeiten in großem Maßstab.
Die Dokumentation verbindet alles, indem sie den Aufsichtsbehörden nachweist, dass die Verfahren tatsächlich eingehalten wurden. In Chargenprotokollen wird festgehalten, was während der Produktion passiert — welche Inspektionen durchgeführt wurden, welche Ergebnisse erzielt wurden, wie Abweichungen behoben wurden und wer die Freigabe genehmigt hat. Bei behördlichen Inspektionen belegen diese Aufzeichnungen, dass Ihr Qualitätskontrollsystem tatsächlich funktioniert und nicht nur als schriftliche Verfahren existiert. Fehlende Unterlagen führen zu Bedenken hinsichtlich der Einhaltung der Vorschriften, auch wenn die Etiketten selbst korrekt sind, da die Aufsichtsbehörden nicht überprüfen können, ob der Prozess wie geplant funktioniert hat. Unternehmen, die diese Qualitätskontrollmaßnahmen vom Entwurf bis zur Dokumentation integrieren, erkennen Kennzeichnungsfehler, bevor sie zu Marktproblemen werden, während Unternehmen, die sich auf reaktive Kontrollen verlassen, Probleme bei Audits entdecken, deren Behebung weitaus teurer ist.
Automatisierungstools für gesetzeskonforme Etikettierung
Diese Plattformen verwalten Arbeitsabläufe, bei denen mehrere Teams die Etiketten vor der Produktion überprüfen, um sicherzustellen, dass die Etikettierung in jeder Phase der Überprüfung konform ist. Die Inhalte werden von der Forschung und Produktentwicklung über die behördliche Prüfung bis hin zur Design- und Marketingphase für das Layout, die Markenkennzeichnung und die Überprüfung der Angaben durch die Behörden weitergeleitet, bevor sie zur endgültigen Überprüfung der Richtigkeit in die Qualitätssicherung gelangen. Das System verfolgt, wer was wann überprüft hat, und erstellt so den Prüfpfad, den behördliche Inspektionen vorschreiben. Unternehmen können diese Workflows über E-Mail-Ketten und gemeinsam genutzte Laufwerke hinaus skalieren.
Die Validierung folgt der gleichen Strenge, die für jedes pharmazeutische Herstellungssystem erforderlich ist. Der Prozess beginnt mit der Installationsqualifizierung zur Bestätigung der korrekten Einrichtung, geht über die Betriebsqualifizierung, bei der alle Funktionen getestet werden, und endet mit einer Leistungsqualifizierung, die die gleichbleibende Genauigkeit im normalen Betrieb nachweist. Die Validierung beweist, dass das automatisierte System Kennzeichnungsfehler zuverlässig verhindert, anstatt neue zu verursachen.
Die Integration mit behördlichen Informationsmanagementsystemen verbindet genehmigte Inhalte direkt mit der Etikettenproduktion. Aktualisierungen der Verschreibungsinformationen nach FDA-Genehmigungen werden direkt auf die entsprechenden Etiketten übertragen, ohne dass jemand manuell Text zwischen den Systemen kopiert. Dadurch werden Übertragungsfehler vermieden und die Änderungszyklen beschleunigt. Zu den Hauptvorteilen gehören schnellere Aktualisierungen der Etiketten und eine strengere Versionskontrolle auf allen globalen Websites, unterstützt durch strukturierte Überprüfungsabläufe, die die allgemeine Einhaltung gesetzlicher Vorschriften verbessern. Unternehmen können ihre betriebliche Effizienz steigern, indem sie manuelle Berührungspunkte reduzieren und den Etikettierungsstatus in allen Fertigungsabläufen in Echtzeit einsehen können.
Bewährte Verfahren für behördliche Einreichungen
Beziehen Sie regulatorische Angelegenheiten in die Produktentwicklung ein, damit Überlegungen zur Kennzeichnung die Planung klinischer Studien und die Datenerhebung beeinflussen können. Wenn Sie bis zur Einreichung warten, müssen Sie nicht weiterkommen und versuchen, Ihre Daten für die Etikettierung geltend zu machen, für deren Nachweis sie nicht konzipiert waren.
Ihre Einreichung muss in dem Format gekennzeichnet werden, das die Agentur erwartet. Die FDA will strukturierte Produktkennzeichnung (SPL) -Format, ein XML-basierter Standard, den ihre Systeme elektronisch verarbeiten können. Der Inhalt entspricht den Industriestandards — die Abschnitte werden in der vorgeschriebenen Reihenfolge angezeigt und enthalten alle erforderlichen Elemente. Wenn Sie die technischen Spezifikationen falsch angeben, wird Ihre Einreichung aufgrund der Formatierung und nicht aufgrund von tatsächlichen Sicherheits- oder Wirksamkeitsbedenken abgelehnt.
Wenn Sie nachverfolgen, was sich gegenüber Ihrer derzeit genehmigten Version geändert hat, sind reibungslose Aktualisierungen der Etikettierung von regulatorischen Problemen getrennt. Einige Änderungen müssen vor der Umsetzung genehmigt werden, andere unterliegen den Bestimmungen, dass Änderungen in Kraft treten, und wieder andere werden nur jährlich gemeldet. Wenn Sie diese kombinieren, entstehen Compliance-Probleme, die die Markteinführung von Updates verzögern. Elektronische Einreichungen haben die Papierform ersetzt, und das Gateway der FDA verarbeitet strukturierte Inhalte anders als PDFs. Viele Unternehmen bieten spezialisierte Dienstleistungen an, um die technischen Anforderungen zu erfüllen und die Genauigkeit sicherzustellen. Nach der Genehmigung müssen Ihre Systeme sicherstellen, dass nur zugelassene Versionen in Produktion gehen. Eine wirklich funktionierende Änderungskontrolle verhindert, dass alte Etiketten nach der Genehmigung neuer Versionen durchrutschen.
Schulungs- und Dokumentationsanforderungen
Jeder, der an der Etikettierung beteiligt ist, muss verstehen, was er tut und warum es wichtig ist, und Sie müssen es beweisen. Bei diesen Sitzungen handelt es sich nicht um einmalige Ereignisse. Kontinuierliche Schulungen halten die Teams über regulatorische Änderungen und neue Systeme auf dem Laufenden. Der eigentliche Maßstab ist nicht, ob Schulungen stattfinden, sondern ob die Teams die aktuellen Anforderungen verstehen, wenn die Inspektoren eintreffen.
Standardarbeitsanweisungen (SOPs) dokumentieren, wie Ihr Unternehmen jeden Schritt des Kennzeichnungsprozesses abwickelt. Sie zeigen den Aufsichtsbehörden, dass Sie einem definierten Prozess folgen, anstatt unterschiedliche Entscheidungen zu treffen, je nachdem, wer an diesem Tag arbeitet. Die SOPs müssen Folgendes abdecken:
- Wie Grafikdateien verwaltet und versionsgesteuert werden
- Wie Genehmigungen innerhalb der Organisation ablaufen
- Wie Änderungen initiiert, überprüft und umgesetzt werden
- Wie Abweichungen und Probleme untersucht und gelöst werden
Das Schreiben von SOPs ist einfach. Sie konsequent zu befolgen und sie zu aktualisieren, wenn sich die Kennzeichnungspraktiken ändern, haben Unternehmen Probleme. Aufzeichnungen belegen, dass Ihr Qualitätssystem wie geplant funktioniert und nicht nur auf dem Papier. Sie müssen nachweisen, dass das geschulte Personal die genehmigten Verfahren befolgt hat, die Inspektionen wie geplant abgeschlossen und alle Abweichungen ordnungsgemäß untersucht wurden. Bei behördlichen Inspektionen beantworten diese Unterlagen die grundlegendste Frage: Haben Sie tatsächlich das getan, was in Ihren Verfahren vorgeschrieben ist? Die Schulung erstreckt sich nicht nur auf Etikettierungsspezialisten, sondern auch auf alle, die mit dem Prozess zu tun haben, von Mitarbeitern für behördliche Angelegenheiten, die Inhalte schreiben, über Grafikdesigner, die Grafiken erstellen, bis hin zum Qualitätssicherungspersonal, das Produktionsläufe genehmigt. Teams, die Wert auf starke Schulungsprogramme legen, haben bei Audits in der Regel weniger Compliance-Probleme und können schneller reagieren, wenn sich Anforderungen ändern. Die Alternative besteht darin, Lücken zu entdecken, wenn die Aufsichtsbehörden sie finden. Ihre Behebung kostet weitaus mehr.
Zukünftige Trends in der Arzneimittelkennzeichnung
Die digitale Etikettierung geht über PDF-Versionen von Papierbeilagen hinaus. QR-Codes auf den Verpackungen sind jetzt direkt mit den Verschreibungsinformationen verknüpft, die in Echtzeit aktualisiert werden, sodass medizinisches Fachpersonal auf aktuelle Sicherheitsdaten zugreifen kann, anstatt sich auf gedruckte Beilagen zu verlassen, die möglicherweise Monate alt sind. Die FDA ist sich bewusst, dass Etiketten auf Papier Grenzen haben, insbesondere für komplexe Produkte, bei denen die umfassenden Informationen den vertretbaren Platzbedarf überschreiten, aber die Aufsichtsbehörden haben die physische Kennzeichnung nicht aufgegeben. Die wichtigsten Sicherheitsinformationen müssen immer noch auf der Verpackung angebracht sein, um den Zugriff ohne digitale Geräte zu gewährleisten.
Der Umweltdruck verändert Verpackungsentscheidungen auf eine Weise, die manchmal im Widerspruch zu behördlichen Anforderungen steht. Die Pharmabranche sieht sich mit der Forderung konfrontiert, Abfall zu reduzieren und nachhaltige Materialien zu verwenden. Ziele, die mit den Vorschriften für bestimmte Verpackungskonstruktionen kollidieren können. Die Europäische Union hat besonders strenge Anforderungen an nachhaltige Verpackungen erlassen, die sich auf die Gestaltung und Produktion von Etiketten auswirken.
Intelligente Etiketten mit NFC-Chips oder eingebetteten Technologien verifizieren die Echtheit der Produkte und verfolgen gleichzeitig die Nutzung und überwachen die Temperaturexposition während des gesamten Vertriebs. Diese Funktionen werfen neue regulatorische Fragen zum Datenschutz und zur Datensicherheit auf und stellen sich auch die Frage, inwieweit Smart-Label-Funktionen bis in den Bereich medizinischer Geräte reichen. Die Technologie ist vorhanden; der regulatorische Rahmen dafür ist noch im Entstehen begriffen.
Ressourcen für Experten für pharmazeutische Etikettierung
Berufsverbände wie die Fachverband für regulatorische Angelegenheiten (RAPS) und die Arzneimittelinformationsverband (DIA) verbindet Sie im Rahmen von Konferenzen und Arbeitsgruppen mit Menschen, die vor ähnlichen Herausforderungen bei der Kennzeichnung stehen. Sie erhalten einen Einblick, wie die Aufsichtsbehörden mit neuen Anforderungen umgehen, und sehen gleichzeitig, wie andere Unternehmen Probleme lösen, mit denen Sie zu tun haben. RAPS bietet auch Zertifizierungen für regulatorische Angelegenheiten an, die sich mit Kennzeichnungskompetenzen befassen. Spezialisierte Schulungsprogramme decken spezifische Lücken ab, wenn Teams sich über neue Vorschriften oder Technologien auf dem Laufenden halten müssen.
Externe Unterstützung kommt aus zwei Richtungen. Beratungsunternehmen verfügen über regulatorisches Fachwissen zur Bewältigung komplexer Anträge, insbesondere wenn Sie es mit unbekannten Anforderungen oder engen Fristen zu tun haben. Technologieanbieter bieten Plattformen an, die das marktübergreifende Versionsmanagement übernehmen und gleichzeitig Änderungen und Genehmigungen koordinieren. Die Wahl zwischen ihnen hängt oft davon ab, ob Ihre Herausforderung darin besteht, zu verstehen, was die Aufsichtsbehörden wollen, oder ob Sie die damit verbundene betriebliche Komplexität bewältigen müssen.
Für Teams, die Inhalte verwalten, bei denen viel auf dem Spiel steht, ist Präzision nicht optional. Verify von GlobalVision gibt den Teams die Gewissheit, dass jede genehmigte Version inspektionsbereit ist.
