

Un flacon mal étiqueté passe tous les points de contrôle et atteint la pharmacie d'un hôpital. Les instructions posologiques semblent routinières : un chiffre de moins que personne ne remarque avant qu'il ne soit trop tard. Cela nous rappelle brutalement que la conformité de l'étiquetage des produits pharmaceutiques demeure l'une des garanties les plus importantes de l'industrie pharmaceutique.
Introduction à la conformité de l'étiquetage des produits pharmaceutiques
L'étiquetage pharmaceutique conforme sert de lien direct entre les sociétés pharmaceutiques et les professionnels de santé, fournissant des informations essentielles qui ont un impact direct sur la sécurité des patients. Cela inclut tout ce qui est visible sur les emballages et les contenants des médicaments, ainsi que le matériel d'accompagnement, afin de garantir que les professionnels de la santé et les patients disposent d'informations précises pour prendre des décisions thérapeutiques sûres et éclairées.
Les organismes de réglementation du monde entier appliquent des directives strictes car les erreurs d'étiquetage peuvent causer de graves dommages. Aux États-Unis, le Administration des aliments et des médicaments (FDA) définit et applique les exigences d'étiquetage des produits pharmaceutiques pour les médicaments sur ordonnance et les dispositifs médicaux. Dans l'ensemble de l'Union européenne, ce rôle incombe au Agence européenne des médicaments (EMA). En coordination avec Santé Canada, Agence japonaise des produits pharmaceutiques et des dispositifs médicaux (PDMA) et Administration des produits thérapeutiques (TGA), ces agences de régulation créent des cadres conçus pour protéger la santé publique et maintenir la cohérence entre les marchés. Efforts visant à harmoniser les normes par le biais du Conseil international pour l'harmonisation (ICH) se poursuivent, mais un alignement complet reste hors de portée, chaque région appliquant ses propres exigences réglementaires que les sociétés pharmaceutiques doivent gérer avec soin. Parallèlement, le paysage réglementaire ne cesse d'évoluer : la diffusion numérique des informations modifie la façon dont les prestataires de soins de santé accèdent aux données, et les mandats de sérialisation redéfinissent le fonctionnement des processus d'étiquetage des produits pharmaceutiques dans l'industrie.
Principaux cadres réglementaires pour l'étiquetage des produits pharmaceutiques
Depuis 2006, la FDA Règle d'étiquetage des médecins (PLR) a défini la norme moderne pour médicament sur ordonnance étiquetage, apportant cohérence et structure à chaque produit approuvé. Il a introduit une hiérarchie claire pour les informations de prescription, avec une section concise « Points forts » destinée aux professionnels de santé et des règles spécifiques concernant la manière dont le contenu restant doit apparaître. Ces réglementations répondent directement aux erreurs de médication qui se produisent lorsque des informations critiques sont enterrées dans un texte dense. Ces réglementations strictes s'appliquent à toutes les catégories de produits, y compris les médicaments en vente libre auxquels les consommateurs ont accès sans ordonnance.
Les normes internationales ajoutent des niveaux d'exigences pour les entreprises opérant sur tous les marchés. L'UE Résumé des caractéristiques du produit (SmPC) et les exigences relatives aux notices diffèrent des formats de la FDA en termes de structure et d'importance accordée au contenu. Alors que les lignes directrices de l'ICH visent à harmoniser les exigences techniques, chaque région applique des approches distinctes. Le tableau ci-dessous montre comment les principaux marchés gèrent différemment les principales exigences en matière d'étiquetage.
Exigences réglementaires en matière d'étiquetage sur les principaux marchés

Le Japon exige que tout le contenu des étiquettes soit rédigé en japonais et mis en forme conformément aux pratiques locales en matière de documentation médicale. La TGA australienne définit ses propres exigences pour le registre australien des produits thérapeutiques, conformément aux normes nationales en matière de clarté et de cohérence. Les entreprises qui lancent des produits pharmaceutiques dans le monde entier doivent conserver plusieurs versions d'étiquettes, chacune répondant aux normes réglementaires locales tout en garantissant la cohérence des principaux messages de sécurité. Gérer contrôle de version l'ensemble de ces marchés nécessite des systèmes permettant de déterminer quelle version d'étiquette s'applique à quel endroit, faisant de la conformité en matière d'étiquetage des produits pharmaceutiques une discipline qui associe expertise en affaires réglementaires et précision opérationnelle pour naviguer dans le labyrinthe réglementaire mondial actuel.
Composantes essentielles des étiquettes conformes des médicaments sur ordonnance
Les étiquettes des médicaments sur ordonnance fournissent des informations différentes selon l'endroit où elles apparaissent. L'étiquette du contenant immédiat, c'est-à-dire le contenu du flacon ou du flacon, couvre les éléments de base : nom du médicament, forme posologique, concentration, voie d'administration et numéro de lot. L'étiquetage des cartons offre plus d'espace de travail. Le panneau d'affichage principal indique le nom et la résistance établis ainsi que la quantité de l'emballage. Les panneaux latéraux contiennent les informations posologiques, les points saillants et les conseils aux patients, ainsi que les informations du fabricant. Les réglementations précisent où chaque élément apparaît et à quel point il est mis en évidence, jusqu'à des tailles de police exactes qui font de l'étiquetage des produits une discipline exigeante.
Il commence par les points saillants qui font apparaître des informations critiques en matière de prescription et de sécurité, puis passe en revue les informations de prescription complètes. Chaque section suit un format prescrit, et tout écart doit être justifié et approuvé par les autorités réglementaires. La structure permet de conserver la cohérence et la facilité de recherche des données de sécurité essentielles, ce qui permet aux examinateurs et aux professionnels de santé d'accéder rapidement aux informations pertinentes. Les sections principales sont les suivantes :
- Indications et utilisation
- Posologie et administration
- Contre-indications
- Avertissements et précautions
- Effets indésirables
- Interactions médicamenteuses
- Utilisation dans des populations spécifiques
- Pharmacologie clinique
Les sections d'information destinées aux patients doivent atteindre un niveau de lecture de la sixième à la huitième année aux États-Unis, comme indiqué dans les directives de lisibilité de la FDA. D'autres agences de régulation peuvent appliquer des normes d'alphabétisation ou de formatage différentes, en fonction de la langue et du contexte sanitaire de leur région. Les guides de médication relatifs aux médicaments présentant des risques graves aident les patients à comprendre comment les utiliser en toute sécurité sans les surcharger de détails techniques. Des instructions d'utilisation et des instructions posologiques claires indiquent aux patients comment prendre correctement leurs médicaments et ce qu'il faut surveiller.
Comprendre le panneau d'information sur les médicaments
Le panneau d'information sur les médicaments fournit aux professionnels de la santé les informations dont ils ont besoin pour prescrire et administrer des médicaments en toute sécurité. Dans ce contenu, les instructions de conservation ont une importance particulière car elles déterminent si un médicament conserve sa stabilité et son efficacité tout au long de sa durée de conservation. Pour protéger cette stabilité, les étiquettes définissent les plages de température requises et expliquent comment protéger les produits de la lumière et de l'humidité pendant le stockage ou le transport. Lorsque ces limites ne sont pas respectées, une dégradation peut survenir avant même qu'un produit n'atteigne les patients, ce qui constitue une perte de qualité évitable qui souligne la nécessité d'un contrôle qualité approfondi. Certains médicaments doivent être réfrigérés, d'autres à l'abri de la lumière, et bon nombre d'entre eux perdent de leur efficacité s'ils sont exposés à l'humidité pendant le transport. Ces exigences déterminent si les produits restent efficaces ou se transforment en substances inutiles (ou nocives). Lorsque ces instructions sont communiquées de manière peu claire ou totalement ignorées, les produits perdent de leur efficacité ou deviennent potentiellement dangereux.
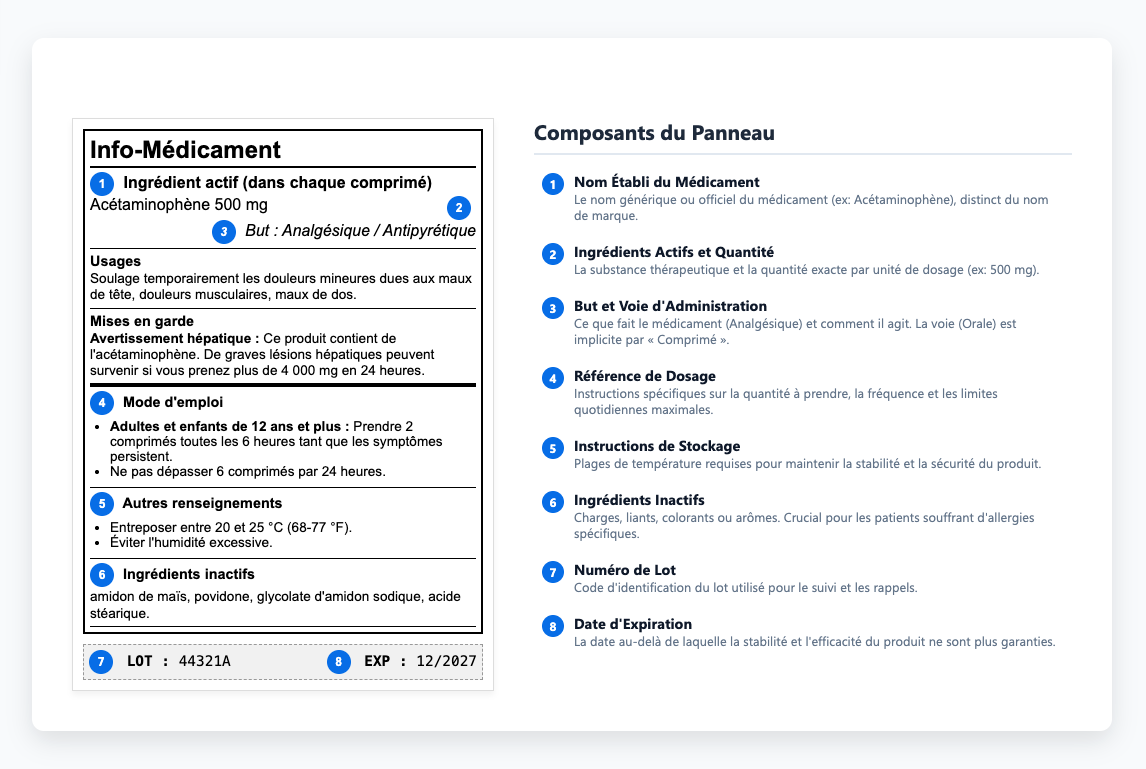
Lorsque les formulations changent ou que de nouvelles données de sécurité apparaissent, les mises à jour des étiquettes sont parfois à la traîne, ce qui crée des incohérences entre les soumissions approuvées et les étiquettes mises sur le marché. Ces incohérences peuvent sembler mineures au premier abord, mais elles apparaissent souvent lors des inspections réglementaires, ce qui ralentit les examens de conformité et ajoute des risques inutiles. Les auditeurs comparent le contenu des étiquettes directement aux versions approuvées, de sorte que ces problèmes restent rarement cachés longtemps.
Étiquettes d'avertissement et informations de sécurité
Les mises en garde en forme de boîte noire constituent la forme d'alerte de sécurité la plus grave de la FDA. Elles sont utilisées pour souligner les risques graves ou potentiellement mortels qui doivent figurer en bonne place sur les étiquettes des médicaments sur ordonnance. Ils apparaissent lorsque les précautions standard ne permettent pas de prévenir des dommages graves. La FDA les ajoute sur la base de données post-commercialisation révélant des schémas d'effets indésirables dangereux, parfois mortels. Ces mises en garde figurent en haut de l'information posologique, dans des bordures en gras, dans un langage direct indiquant le risque et les personnes les plus vulnérables. Les contre-indications vont encore plus loin en identifiant les cas où le médicament ne doit absolument pas être utilisé. Cela inclut les interactions dangereuses avec d'autres médicaments, les risques de grossesse documentés et les problèmes médicaux préexistants qui rendraient le médicament dangereux.
Les informations sur les effets indésirables couvrent les effets légers attendus jusqu'aux complications rares mais graves. La présentation des données de fréquence parallèlement à ces résultats permet de situer le niveau de risque dans son contexte pour les patients et les évaluateurs. Pour réduire ce risque, les stratégies d'atténuation consistent souvent à ajuster les doses pour des populations spécifiques, à appliquer des protocoles de surveillance et à orienter les prestataires de soins de santé sur les conseils et le suivi appropriés. Pour certains médicaments à haut risque, Stratégies d'évaluation et d'atténuation des risques Les programmes (REMS) prescrivent des éléments d'étiquetage supplémentaires pour garantir la sécurité des patients en tant qu'exigences contraignantes.

Exigences d'étiquetage spécialisées pour les médicaments génériques
L'étiquetage des médicaments génériques doit correspondre presque exactement à l'étiquette du produit de marque, une exigence que la FDA applique pour garantir que les génériques offrent la même sécurité et la même efficacité que l'original. Vous pouvez différer sur certains points : les informations sur les brevets sont omises car elles appartiennent à la marque, tandis que les informations relatives au fabricant et les codes NDC remplacent les leurs. La section « Mode d'approvisionnement » peut également refléter votre emballage spécifique.
Le véritable travail commence lorsque l'étiquette de la marque change. Une nouvelle mise en garde ou une nouvelle contre-indication est ajoutée sur la base des données post-commercialisation, et votre étiquette générique doit correspondre. La procédure « Changements en cours » de la FDA vous permet d'effectuer certaines mises à jour de sécurité sans attendre l'approbation, mais vous êtes toujours responsable de détecter ces changements et d'y donner suite. Les entreprises qui comptent sur les régulateurs pour les informer lorsque les étiquettes sont obsolètes finissent par prendre du retard, réglant les problèmes une fois que les inspections réglementaires les ont détectées au lieu de garder une longueur d'avance. Pour rester à jour, vous devez suivre les modifications apportées aux produits de référence, puis évaluer l'impact de ces mises à jour sur vos produits spécifiques. Les mises à jour doivent être déployées sur tous les sites de fabrication avec une documentation prouvant la conformité lors des audits. La pression augmente lorsque les mises à jour de sécurité nécessitent une action immédiate alors que vos processus normaux prennent des mois. Les fabricants de médicaments génériques qui gèrent cette situation bien mettent au point des systèmes de surveillance qui détectent les changements rapidement au lieu d'attendre que des problèmes apparaissent lors des examens de conformité de l'étiquetage.
Produits biologiques : considérations spécifiques en matière d'étiquetage
Les produits biologiques proviennent d'organismes vivants et non de la synthèse chimique, qui modifie ce qui doit figurer sur les étiquettes. Les produits biologiques nécessitent des informations sur leur source biologique et leurs procédés de fabrication, ainsi que des avertissements concernant les réponses du système immunitaire que les médicaments chimiques ne déclenchent pas.
Principales différences d'étiquetage
Les exigences de stockage des produits biologiques vont au-delà des spécifications standard, car ces produits se dégradent facilement dans des conditions qui n'affecteraient pas les médicaments chimiques. La plupart ont besoin d'être réfrigérés dans des plages de température étroites, et une brève exposition à la chaleur, à la lumière ou à l'agitation peut les rendre inefficaces ou potentiellement nocifs. Les étiquettes indiquent comment les produits doivent être stockés et transportés, en détaillant les limites de température et les étapes de manipulation confirmées au cours du développement.
Les mises en garde relatives à l'immunogénicité expliquent comment le système immunitaire peut réagir aux produits biologiques. Les étiquettes expliquent le risque de développer des anticorps antimédicamenteux et résument la manière dont les essais cliniques ont évalué ce risque, y compris ce que les résultats signifient pour les patients. Les professionnels de santé utilisent ces informations pour surveiller la réponse au traitement et ajuster le traitement en cas de réactions immunitaires. La surveillance après la mise sur le marché va au-delà de la notification standard des effets indésirables, avec des étiquettes spécifiant les exigences de participation au registre ou une surveillance à long terme qui permet de suivre les effets que les essais cliniques n'ont pas pu pleinement saisir.
Défis et solutions en matière d'étiquetage multilingue
Les sociétés pharmaceutiques actives dans le monde entier sont confrontées à des défis particuliers lorsqu'il s'agit de créer des étiquettes qui répondent à diverses exigences linguistiques tout en préservant la précision et la conformité réglementaire.
Défi : qualité de traduction et précision culturelle
La terminologie médicale ne se traduit pas directement d'une langue à l'autre, et le contexte culturel façonne la façon dont les patients comprennent les informations de santé. Une traduction techniquement précise peut complètement passer à côté de la façon dont les personnes d'un marché cible abordent réellement les concepts médicaux. Les services de traduction standard ne disposent pas des connaissances spécialisées requises en matière d'étiquetage des produits pharmaceutiques.
Une traduction efficace nécessite des spécialistes qui comprennent à la fois la terminologie médicale et les attentes réglementaires de chaque marché cible. Les traducteurs ont besoin d'une maîtrise native et d'une connaissance pratique du domaine pharmaceutique pour garantir l'exactitude et le contexte. La FDA et d'autres agences de réglementation indiquent dans quels cas les documents traduits nécessitent une approbation officielle ou un traducteur certifié, avec des règles spécifiques qui varient selon les régions.
Défi : répondre aux diverses exigences linguistiques du marché
De nombreux marchés exigent la présence de plusieurs langues sur une seule étiquette. Le Canada exige le bilinguisme en français et en anglais. La Suisse a besoin de l'allemand, du français et de l'italien. La Belgique exige le français, le néerlandais et l'allemand. L'insertion de plusieurs traductions complètes sur des étiquettes dont l'espace est restreint impose de difficiles compromis entre lisibilité et respect des exigences réglementaires. Les entreprises y remédient en collaborant avec des concepteurs d'étiquettes spécialisés dans les emballages pharmaceutiques multilingues, en utilisant des techniques telles que le pliage en accordéon, les étiquettes de brochures ou les hiérarchies d'informations hiérarchisées qui donnent la priorité aux informations de sécurité critiques.
Défi : Coordonner les mises à jour entre les langues et les marchés
Lorsque les informations de sécurité sont modifiées sur l'étiquette principale, chaque version traduite doit être mise à jour. Les délais réglementaires sur les différents marchés s'alignent rarement, ce qui crée des périodes pendant lesquelles différents pays affichent des informations de sécurité différentes sur leurs étiquettes.
La technologie fournit des solutions dans ce domaine. Les systèmes de mémoires de traduction assurent la cohérence terminologique entre les produits, tandis que les bases de données terminologiques garantissent une traduction uniforme des termes techniques à chaque fois qu'ils apparaissent. Les systèmes de gestion des flux de travail réunissent tout cela en suivant quelles versions d'étiquettes existent dans quelles langues, quels marchés ont approuvé chaque version et où des mises à jour sont nécessaires. Ces systèmes aident les entreprises à maintenir leur conformité en signalant les lacunes et en coordonnant le contrôle des versions entre les opérations mondiales avant que les inspections réglementaires ne révèlent des problèmes.
Exigences en matière de sérialisation et de suivi et de traçabilité
Les réglementations relatives à la sérialisation ont modifié le fonctionnement de l'étiquetage des produits pharmaceutiques en exigeant que chaque emballage porte un identifiant unique permettant de le suivre tout au long de la chaîne d'approvisionnement. Cela permet de résoudre de véritables problèmes, notamment la contrefaçon et la nécessité de procéder à des rappels rapides lorsque des problèmes de sécurité apparaissent. Les réglementations du monde entier imposent désormais la sérialisation, bien que les délais de mise en œuvre varient selon les marchés. La loi américaine sur la sécurité de la chaîne d'approvisionnement en médicaments exigeait la conformité d'ici 2023, tandis que la directive européenne sur les médicaments falsifiés est entrée en vigueur en 2019. D'autres marchés ont suivi leur propre calendrier, ce qui a entraîné un déploiement mondial échelonné qui a obligé les entreprises à gérer différentes exigences simultanément. Ces réglementations exigent que les emballages comportent des identificateurs de produits comportant de multiples éléments de données (codes nationaux des médicaments et numéros de série uniques ainsi que numéros de lot et dates de péremption), le tout dans des formats lisibles par les humains et les machines.
La technologie repose principalement sur les codes-barres 2D Data Matrix, qui stockent de grandes quantités d'informations dans un format compact. Ces codes apparaissent à chaque étape de la production, des chaînes de fabrication aux systèmes de contrôle qualité en passant par les bases de données de distribution qui retracent chaque identifiant unique. Pour la plupart des équipes, la véritable complexité réside dans le fait que ces exigences doivent coexister avec des étiquettes contenant déjà du contenu obligatoire. Les codes-barres ne peuvent pas masquer le texte requis et les numéros de série doivent apparaître dans des formats lisibles à la fois par l'homme et par machine. Dans le même temps, les données de sérialisation doivent rester dans leur position traditionnelle pour être conformes aux processus d'étiquetage des produits pharmaceutiques de longue date. Dans la pratique, les entreprises conçoivent désormais des étiquettes qui répondent à deux générations d'attentes réglementaires tout en préservant la clarté et la lisibilité.
Contrôle de la qualité dans l'étiquetage des produits pharmaceutiques
Les examens de conception des étiquettes comparent les étiquettes proposées aux soumissions réglementaires approuvées. Le texte doit correspondre exactement, les graphiques doivent apparaître comme approuvés, la mise en forme doit suivre les spécifications. Même les écarts mineurs nécessitent une documentation et déclenchent souvent une notification réglementaire. C'est pourquoi ces examens constituent la base de référence pour toutes les vérifications ultérieures. Une fois les preuves fournies par les fournisseurs, les outils d'inspection alimentés par l'IA aident les équipes d'assurance qualité entrantes à vérifier que les fichiers correspondent aux documents maîtres approuvés avant le début de l'impression :
- Comparaison d'œuvres d'art vérifie les preuves par rapport aux modèles approuvés pour détecter les changements de mise en page ou les éléments manquants
- Vérification des codes-barres et des codes QR confirme que les données codées sont correctes et que chaque code suit le formatage requis avant l'impression
- Reconnaissance optique de caractères (OCR) identifie les différences de texte entre les épreuves et les fichiers maîtres approuvés afin de garantir qu'aucune modification involontaire n'a été introduite lors des mises à jour
- Détection des modifications prévues vérifie si les corrections annotées des versions précédentes des fichiers ont été appliquées correctement, aidant ainsi les équipes à confirmer que les mises à jour demandées ont été mises en œuvre avant la production.
Ces systèmes fonctionnent en continu pendant les cycles de production, analysant les illustrations et le texte pour signaler immédiatement les problèmes plutôt que de les découvrir après l'impression de milliers d'unités. La supervision humaine gère les exceptions et fournit l'approbation finale, mais l'inspection automatisée assure l'essentiel du travail de vérification à grande échelle.
La documentation permet de tout relier en prouvant aux régulateurs que les procédures ont été effectivement suivies. Les dossiers de lots enregistrent ce qui se passe pendant la production : quelles inspections ont été effectuées, quels résultats ont été obtenus, comment les écarts ont été résolus, qui a approuvé la libération. Lors des inspections réglementaires, ces enregistrements démontrent que votre système de contrôle qualité fonctionne réellement au lieu de simplement exister sous forme de procédures écrites. L'absence de documentation pose des problèmes de conformité, même lorsque les étiquettes elles-mêmes sont correctes, car les régulateurs ne peuvent pas vérifier que le processus a fonctionné comme prévu. Les entreprises qui intègrent ces mesures de contrôle qualité depuis la conception jusqu'à la documentation détectent les erreurs d'étiquetage avant qu'elles ne deviennent des problèmes de marché, tandis que celles qui s'appuient sur des contrôles réactifs découvrent les problèmes lors des audits lorsque les réparations sont beaucoup plus coûteuses.
Outils d'automatisation pour un étiquetage conforme
Ces plateformes gèrent les flux de travail dans lesquels plusieurs équipes révisent les étiquettes avant la production, garantissant ainsi un étiquetage conforme à chaque étape de la révision. Le contenu passe de la recherche et du développement de produits à l'examen réglementaire, puis aux étapes de conception et de marketing pour la mise en page, l'image de marque et la vérification des allégations avant d'être soumis à l'assurance qualité pour les derniers contrôles d'exactitude. Le système permet de suivre qui a examiné quoi et quand, créant ainsi la piste d'audit exigée par les inspections réglementaires. Les entreprises peuvent étendre ces flux de travail au-delà de ce que les chaînes de messagerie et les lecteurs partagés peuvent gérer.
La validation suit la même rigueur que celle requise pour tout système de fabrication pharmaceutique. Le processus commence par la qualification de l'installation pour confirmer la configuration correcte, passe par une qualification opérationnelle qui teste toutes les fonctions et se termine par une qualification des performances démontrant une précision constante pendant les opérations normales. La validation prouve que le système automatisé prévient de manière fiable les erreurs d'étiquetage au lieu d'en introduire de nouvelles.
L'intégration aux systèmes de gestion des informations réglementaires connecte le contenu approuvé directement à la production d'étiquettes. Les mises à jour des informations de prescription après les approbations de la FDA sont directement répercutées sur les étiquettes pertinentes sans que personne ne copie manuellement le texte entre les systèmes, ce qui réduit les erreurs de transcription tout en accélérant les cycles de modification. Les principaux avantages incluent des mises à jour plus rapides des étiquettes et un contrôle des versions plus strict sur les sites mondiaux, soutenus par des flux de révision structurés qui renforcent la conformité réglementaire globale. Les entreprises constatent des gains d'efficacité opérationnelle en réduisant les points de contact manuels et en bénéficiant d'une visibilité en temps réel sur l'état de l'étiquetage dans l'ensemble des opérations de fabrication.
Meilleures pratiques pour les soumissions réglementaires
Impliquez les affaires réglementaires lors du développement des produits afin que les considérations relatives à l'étiquetage puissent influencer la manière dont vous concevez les essais cliniques et collectez les données. Si vous attendez le délai de dépôt, vous êtes bloqué à essayer de faire en sorte que vos données puissent étayer des allégations sur l'étiquetage pour lesquelles elles n'ont pas été conçues pour prouver.
Votre soumission doit être étiquetée dans le format attendu par l'agence. La FDA veut étiquetage structuré des produits (SPL), une norme basée sur XML que leurs systèmes peuvent traiter électroniquement. Le contenu est conforme aux normes de l'industrie : les sections apparaissent dans l'ordre prescrit avec tous les éléments requis inclus. Si vous vous trompez dans les spécifications techniques, votre demande sera rejetée pour des raisons de formatage plutôt que pour des raisons de sécurité ou d'efficacité réelles.
Le suivi des modifications par rapport à votre version actuellement approuvée permet de distinguer les mises à jour fluides de l'étiquetage des problèmes réglementaires. Certains changements doivent être approuvés au préalable avant d'être mis en œuvre, d'autres sont traités dans le cadre des dispositions relatives aux « modifications en cours d'application », et d'autres font simplement l'objet d'un rapport annuel. Si vous les mélangez, vous créez des problèmes de conformité qui ralentissent la mise sur le marché des mises à jour. Les soumissions électroniques ont remplacé le papier, le Gateway de la FDA traitant le contenu structuré différemment des PDF. De nombreuses entreprises font appel à des services spécialisés pour répondre aux exigences techniques et garantir la précision. Après approbation, vos systèmes doivent garantir que seules les versions approuvées seront mises en production. Un contrôle des modifications qui fonctionne réellement empêche les anciennes étiquettes de s'infiltrer une fois les nouvelles versions approuvées.
Exigences en matière de formation et de documentation
Toutes les personnes impliquées dans les opérations d'étiquetage doivent comprendre ce qu'elles font et pourquoi c'est important, et vous devez le prouver. Ces sessions ne sont pas des événements ponctuels. La formation continue permet aux équipes de rester informées des changements réglementaires et des nouveaux systèmes. La véritable mesure n'est pas de savoir si la formation est dispensée, mais de savoir si les équipes comprennent les exigences actuelles à l'arrivée des inspecteurs.
Les procédures opérationnelles standard (SOP) documentent la manière dont votre entreprise gère chaque étape du processus d'étiquetage, en montrant aux régulateurs que vous suivez un processus défini plutôt que de prendre des décisions différemment en fonction de la personne qui travaille ce jour-là. Les SOP doivent couvrir :
- Comment les fichiers d'illustrations sont gérés et leur version contrôlée
- Comment les approbations circulent dans l'organisation
- Comment les changements sont initiés, révisés et mis en œuvre
- Comment les écarts et les problèmes sont étudiés et résolus
La rédaction des SOP est simple ; les entreprises ont du mal à les suivre régulièrement et à les mettre à jour lorsque les pratiques d'étiquetage changent. Les dossiers montrent que votre système qualité fonctionne comme prévu, et pas seulement sur papier. Ils doivent démontrer que le personnel qualifié a suivi les procédures approuvées, que les inspections ont été effectuées comme prévu et que tout écart a fait l'objet d'une enquête appropriée. Lors des inspections réglementaires, ces fichiers répondent à la question la plus fondamentale : avez-vous réellement suivi les instructions de vos procédures ? La formation s'étend au-delà des spécialistes de l'étiquetage et s'adresse à tous ceux qui sont concernés par le processus, qu'il s'agisse du personnel des affaires réglementaires qui rédige du contenu, des graphistes qui créent des illustrations ou du personnel d'assurance qualité qui approuve les cycles de production. Les équipes qui mettent l'accent sur de solides programmes de formation ont tendance à rencontrer moins de problèmes de conformité lors des audits et peuvent réagir plus rapidement lorsque les exigences changent. L'alternative est de découvrir les lacunes lorsque les régulateurs les détectent, ce qui coûte beaucoup plus cher à corriger.
Tendances futures en matière d'étiquetage des produits pharmaceutiques
L'étiquetage numérique va au-delà des versions PDF des encarts papier. Les codes QR figurant sur les emballages renvoient désormais directement aux informations de prescription mises à jour en temps réel, ce qui permet aux professionnels de santé d'accéder aux données de sécurité actuelles plutôt que de se fier à des encarts imprimés datant de plusieurs mois. La FDA reconnaît que les étiquettes en papier ont des limites, en particulier pour les produits complexes pour lesquels des informations complètes dépassent les contraintes d'espace raisonnables, mais les régulateurs n'ont pas abandonné l'étiquetage physique. Les informations de sécurité de base doivent toujours figurer sur l'emballage pour garantir l'accès sans appareils numériques.
La pression environnementale redéfinit les décisions en matière d'emballage d'une manière qui entre parfois en conflit avec les exigences réglementaires. Le secteur pharmaceutique est confronté à des exigences de réduction des déchets et d'utilisation de matériaux durables, des objectifs qui peuvent entrer en conflit avec les réglementations spécifiant des constructions d'emballage particulières. L'Union européenne a imposé des exigences d'emballage durables particulièrement strictes qui affectent la conception et la production des étiquettes.
Les étiquettes intelligentes dotées de puces NFC ou de technologies intégrées vérifient l'authenticité du produit tout en suivant son utilisation et en surveillant l'exposition à la température tout au long de la distribution. Ces fonctionnalités soulèvent de nouvelles questions réglementaires concernant la confidentialité et la sécurité des données, ainsi que la mesure dans laquelle la fonctionnalité d'étiquetage intelligent s'étend au territoire des dispositifs médicaux. La technologie existe ; son cadre réglementaire est encore en train de se former.
Ressources pour les professionnels de l'étiquetage des produits pharmaceutiques
Des associations professionnelles telles que Société des professionnels des affaires réglementaires (RAPS) et le Association pour l'information sur les médicaments (DIA) vous met en contact avec des personnes confrontées à des défis similaires en matière d'étiquetage par le biais de conférences et de groupes de travail. Vous obtenez un aperçu de la manière dont les régulateurs abordent les nouvelles exigences tout en observant comment d'autres entreprises résolvent les problèmes auxquels vous êtes confrontés. Le RAPS propose également une certification en affaires réglementaires qui couvre les compétences en matière d'étiquetage, tandis que des programmes de formation spécialisés comblent des lacunes spécifiques lorsque les équipes doivent se tenir au courant des nouvelles réglementations ou technologies.
Le soutien externe provient de deux directions. Les sociétés de conseil apportent leur expertise en matière de réglementation pour gérer des demandes complexes, en particulier lorsque vous devez faire face à des exigences inconnues ou à des délais serrés. Les fournisseurs de technologies proposent des plateformes qui gèrent la gestion des versions sur tous les marchés tout en coordonnant les modifications et les approbations. Le choix entre les deux dépend souvent du défi que vous devez relever : comprendre ce que veulent les régulateurs ou gérer la complexité opérationnelle liée à leur mise en œuvre.
Pour les équipes qui gèrent du contenu à enjeux élevés, la précision n'est pas facultative. Vérification de GlobalVision donne aux équipes l'assurance que chaque version approuvée est prête à être inspectée.
