
Une étiquette XML égarée peut renvoyer l'intégralité d'une demande de médicament pour correction. Les systèmes automatisés de la FDA rejettent instantanément les fichiers lorsque le formatage ne correspond pas aux exigences du schéma, quelle que soit l'expérience de votre équipe de réglementation.
Introduction à l'étiquetage structuré des produits (SPL)
La FDA oblige les fabricants de produits pharmaceutiques et de dispositifs médicaux à soumettre des informations sur les produits et les installations en utilisant une norme de balisage des documents approuvée pour les soumissions réglementaires. La norme, connue sous le nom de Étiquetage structuré des produits (SPL), utilise un langage de balisage extensible (XML) pour transformer les informations de prescription en données lisibles par machine. Basé sur le cadre Health Level Seven (HL7) utilisé par les agences de réglementation mondiales, le logiciel SPL redéfinit la façon dont les équipes réglementaires préparent les soumissions. Grâce aux spécifications techniques gérées par le système, les professionnels de la réglementation peuvent se concentrer sur la précision du contenu et la stratégie de conformité qui protègent les patients et améliorent les délais de mise sur le marché.
Pourquoi des données produits structurées plutôt que des PDF ? Parce que les autorités réglementaires ont besoin d'extraire des informations sur des milliers de produits pharmaceutiques, puis d'analyser les modèles et de croiser des détails qui seraient invisibles dans des documents non structurés. Lorsque l'étiquetage de votre produit est dans un format standard, les systèmes de la FDA peuvent automatiquement vérifier que votre code national des médicaments correspond à l'enregistrement de votre établissement lors des processus d'inscription des médicaments. Les contrôles automatisés signalent les incohérences, mais les équipes réglementaires continuent d'examiner et de résoudre les divergences avant de les soumettre. Les configurations d'emballage sont suivies par tous les fabricants, tandis que les signaux de sécurité apparaissent plus rapidement lors des examens de pharmacovigilance.
La création manuelle de SPL oblige les équipes chargées des affaires réglementaires à coder manuellement le code XML et à recouper constamment les règles des schémas. Cette approche a fonctionné lorsque les volumes de soumissions étaient plus faibles. Désormais, grâce à des délais d'approbation accélérés et à des mises à jour continues de l'étiquetage, la procédure manuelle introduit des erreurs que les systèmes automatisés détectent instantanément. Alors que le logiciel SPL peut valider automatiquement le schéma et croiser des éléments clés, tels que les codes nationaux des médicaments et les enregistrements des établissements, le personnel de réglementation continue à examiner les divergences signalées pour garantir l'exactitude du contenu avant la soumission.
Comprendre les exigences et les normes SPL
Même une seule erreur de schéma entraîne un rejet de la FDA. Les règles de validation sont strictes et non facultatives, et vos soumissions doivent correspondre exactement à celles-ci. Les fichiers de validation comparent chaque soumission XML aux exigences SPL actuelles afin d'identifier les erreurs structurelles avant qu'elle ne soit envoyée à la FDA. Pour les équipes réglementaires, la mise en place d'un schéma correct signifie simplement éviter les erreurs de formatage à l'origine des rejets. En pratique, cela implique de comprendre quelques composants fondamentaux du SPL :
- Les types de documents indiquent à quelle catégorie appartient une soumission. Les exemples incluent l'enregistrement des établissements, la liste des médicaments ou le contenu de l'étiquetage.
- Les ID de jeu et les ID de version assurent la continuité des documents, en reliant chaque mise à jour à sa version précédente tout au long du cycle de vie du produit.
- Les noms et codes d'identification logiques d'observation (LOINC) aident la FDA et les soumissionnaires à localiser et à valider des sections de contenu spécifiques dans les fichiers SPL, en particulier pour l'étiquetage et les données cliniques, garantissant ainsi une analyse et un traitement précis des documents.
- Chaque fichier SPL définit les principaux éléments de données du produit, de la forme posologique et de la concentration à la configuration de l'emballage, garantissant ainsi une représentation cohérente de chaque composant.
- Les règles standard de balisage des documents définissent la structure des éléments et les vocabulaires contrôlés requis pour les noms des ingrédients.
La norme SPL ne reste pas statique. La FDA publie des schémas mis à jour ainsi que des règles de validation révisées, et des documents d'orientation suivent pour clarifier les attentes en matière de soumission. En suivant le rythme manuellement, votre équipe surveille les annonces réglementaires tout en mettant à jour les modèles internes. Ensuite, vous revalidez vos processus chaque fois que les spécifications changent. Cette charge de maintenance augmente à mesure que votre portefeuille de produits s'agrandit. C'est pourquoi la plupart des équipes réglementaires finissent par abandonner complètement les processus manuels.
Avantages de l'utilisation des solutions logicielles SPL
Les équipes constatent des gains d'efficacité presque immédiatement après l'adoption du logiciel SPL. Le système convertit les documents en XML conforme, valide le schéma et la structure en temps réel et produit des rapports prêts pour l'audit avant leur soumission. Alors que l'automatisation gère les contrôles techniques, la précision du contenu et l'interprétation des réglementations dépendent toujours de l'expertise humaine. Cette validation intégrée permet d'éviter les rejets techniques susceptibles de retarder l'examen réglementaire et de prolonger les délais d'approbation. Les incohérences de schéma qui entraîneraient des rejets de la FDA sont signalées instantanément, de sorte que vous savez que votre soumission sera acceptée avant de l'envoyer.
Le logiciel SPL offre aux équipes réglementaires un espace unique et organisé pour gérer les données d'étiquetage et contrôler les versions au fur et à mesure de l'évolution des produits. Chaque modification est enregistrée automatiquement et les soumissions précédentes restent liées à leurs documents sources, de sorte que l'historique complet est facile à suivre. Lorsque les auditeurs demandent des informations sur une mise à jour précédente, le personnel de réglementation peut ouvrir l'enregistrement directement dans le système. La documentation est associée à l'historique des modifications, de sorte que la récupération ne prend que quelques minutes au lieu de plusieurs heures. Au fil du temps, la préparation de l'audit devient un travail de routine plutôt qu'une tâche stressante de dernière minute.
Les systèmes SPL s'intègrent également à l'écosystème technologique réglementaire plus large. Ils complètent les plateformes existantes pour la gestion des illustrations, l'étiquetage, la création de contenu et la gestion des informations réglementaires, contribuant à créer un flux continu de données approuvées, de la création du contenu à la soumission finale. Cette configuration connectée réduit les doublons et les transferts manuels de données tout en maintenant l'alignement des équipes chargées de la qualité, de la réglementation et de l'étiquetage tout au long du processus.

Principales caractéristiques du logiciel SPL moderne
Le logiciel SPL moderne gère automatiquement la conversion XML et la validation en temps réel, supprimant ainsi une grande partie du codage manuel qui provoquait des erreurs de stade avancé. De nombreuses plateformes se connectent désormais aux systèmes de gestion des informations réglementaires et utilisent des interfaces qui accélèrent et simplifient le travail de soumission pour les équipes réglementaires dans des délais serrés. Sans ces fonctionnalités, les équipes finissent par s'appuyer sur des solutions de contournement qui ralentissent les évaluations et présentent des risques. La bonne plateforme amplifie l'expertise en gérant automatiquement les tâches techniques, ce qui permet aux professionnels de la réglementation de se concentrer sur les décisions qui nécessitent réellement une vision humaine.
Types de modèles de déploiement de logiciels SPL
SaaS ou sur site ? Ce choix détermine la rapidité avec laquelle vous allez soumettre votre demande et qui gère les mises à jour lorsque la FDA modifie ses exigences. Le bon modèle dépend du volume de vos soumissions et de la nécessité d'un contrôle strict des données.
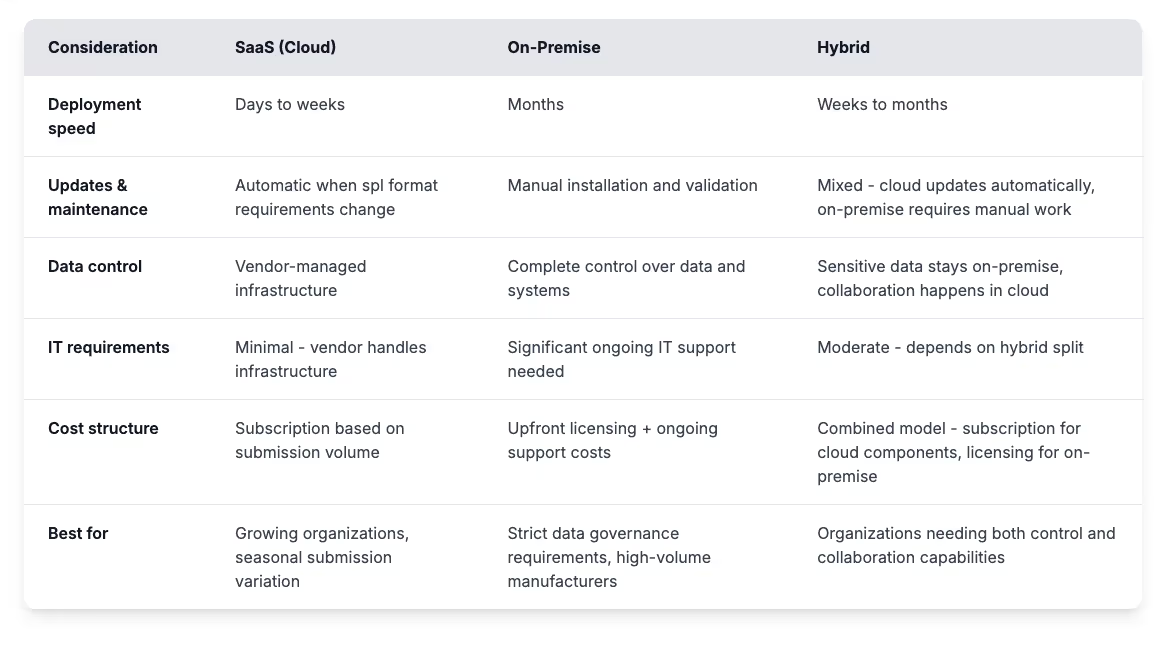
Le SaaS offre des coûts prévisibles à court terme ; les modèles sur site nécessitent des dépenses initiales plus élevées mais peuvent être plus économiques à grande échelle si vous disposez de ressources informatiques pour gérer les mises à jour. Sélectionnez le modèle de déploiement qui équilibre la fréquence de vos envois avec le niveau de contrôle des données et de support informatique que votre équipe peut supporter.
Logiciel SPL pour différents secteurs de l'industrie
Les équipes pharmaceutiques s'appuient sur les plateformes SPL pour certaines fonctions essentielles qui garantissent la cohérence des informations d'étiquetage. Dans ces systèmes, les rapports de distribution des lots permettent de suivre chaque lot, et les données relatives aux événements indésirables sont reconnectées à la version d'étiquetage correspondante dans l'enregistrement de soumission. Ensemble, ces éléments constituent la base de la plupart des plateformes SPL, bien que chaque secteur les utilise un peu différemment.
Pour les produits biologiques, le processus se complique. Un changement dans la fabrication, en particulier dans les systèmes de culture cellulaire, peut entraîner de nouvelles exigences d'étiquetage que les produits à petites molécules rencontrent rarement. Cette évolution modifie également les attentes de la FDA en matière d'informations sur les installations, obligeant les systèmes à gérer les données biologiques avec plus de flexibilité. Les meilleurs outils SPL s'adaptent à ces nuances au lieu de limiter les utilisateurs à des flux de travail spécifiques aux produits pharmaceutiques.
Les fabricants de cosmétiques sont soumis à une surveillance réglementaire moins stricte que les entreprises pharmaceutiques ou de dispositifs, mais les processus d'étiquetage standardisés apportent toujours une valeur ajoutée lorsque les gammes de produits couvrent plusieurs marchés. Ils ne nécessitent peut-être pas la même profondeur de suivi de validation ou d'intégration des événements indésirables, mais des flux de travail cohérents améliorent l'efficacité et réduisent les risques d'erreur.
Beyond SPL : logiciel de soumission réglementaire associé
Les soumissions SPL se connectent directement à des systèmes réglementaires plus larges via Document technique commun électronique intégration (eCTD), étant donné que l'étiquetage existe rarement de manière isolée.
Les rapports de sécurité sont souvent liés au SPL de manière pratique. Par exemple, les soumissions de stratégie d'évaluation et d'atténuation des risques (REMS) peuvent utiliser le format SPL, qui permet aux organisations de gérer des programmes de distribution restreinte dans le même système que celui utilisé pour l'étiquetage standard. Les fonctionnalités de rapport individuel de sécurité des cas (ICSR) facilitent le suivi des effets indésirables, bien que les mises à jour de l'étiquetage basées sur les données de sécurité nécessitent toujours un examen manuel et une prise de décision réglementaire plutôt qu'une synchronisation automatique.
Les soumissions transfrontalières étendent encore ces fonctionnalités. Monographie de produit structurée de Santé Canada (SPM) partage une base technique avec la norme SPL de la FDA, mais inclut des exigences supplémentaires telles que des déclarations en langage clair et des ensembles de codes spécifiques à chaque pays pour répondre aux besoins réglementaires canadiens. Dans la pratique, cela signifie que les fabricants de produits biologiques peuvent gérer les deux marchés à l'aide de systèmes compatibles SPL similaires, en s'appuyant sur les rapports de distribution des lots pour suivre la destination de chaque lot de produits. Lorsque ces types de soumission adjacents se connectent à SPL, les transferts manuels disparaissent, tout comme la plupart des erreurs qui les accompagnent.
Services SPL externalisés par rapport aux logiciels internes
Plusieurs plateformes prennent désormais en charge la création et la gestion de SPL de différentes manières. Le portail SPL d'Intagras fournit un environnement basé sur le cloud avec une validation en temps réel, tandis que Thinspring se concentre sur la création de contenu structuré couvrant plusieurs types de soumissions auprès de la FDA. Les fabricants d'appareils utilisent souvent des systèmes tels que Reed Tech, qui peuvent gérer les soumissions UDI ainsi que les fichiers SPL. Du côté des services, des organisations telles que Freyr SPL SPM et IQVIA gèrent des projets de conversion complets pour les entreprises confrontées à des données de produits complexes ou à des délais d'exécution courts. La collaboration avec ces fournisseurs permet aux équipes chargées des affaires réglementaires de rester concentrées sur la stratégie et la supervision de la conformité plutôt que sur le formatage technique des documents.
Lorsque les volumes de soumissions sont plus élevés, les logiciels deviennent généralement plus économiques, car les frais de service récurrents s'accumulent sur plusieurs années. Les organisations dont la charge de travail réglementaire est plus légère trouvent souvent que l'externalisation est plus rentable. Les fournisseurs de services maîtrisent la complexité liée à la mise en conformité avec les changements de format sans que vous ayez à développer une expertise interne.
Quelle approche répond à vos besoins ?

Ces catégories constituent la base de la décision, mais de nombreuses organisations estiment qu'elles n'ont pas besoin d'en choisir une seule. Les logiciels peuvent gérer les soumissions SPL de routine pendant que les fournisseurs de services interviennent pour des conversions complexes ou des tâches surchargées dans des délais serrés. Cette flexibilité hybride est importante lorsque les demandes de soumission fluctuent de façon imprévisible.
Quelle que soit l'approche, la formation et le soutien façonnent la mise en œuvre. L'utilisation d'un logiciel interne signifie que les équipes doivent comprendre comment les flux de travail de soumission et les processus de validation s'intègrent, car le dépannage fait rapidement partie du travail quotidien. De nombreux professionnels de la réglementation apprennent mieux grâce à une intégration structurée et à une assistance constante aux utilisateurs, mais il faut encore du temps pour acquérir de véritables compétences. Les services externalisés suppriment une grande partie de cette courbe d'apprentissage, car les fournisseurs gèrent le processus technique tandis que votre équipe se concentre sur la précision du contenu et la stratégie réglementaire.
Meilleures pratiques de mise en œuvre
Les implémentations logicielles SPL réussies suivent un déploiement progressif. Le déploiement par étapes protège les flux de soumission et garantit que la validation et la formation restent cohérentes.
Étape 1 : Commencez par un projet pilote ciblé.
Commencez par une gamme de produits ou un domaine thérapeutique avant de vous étendre à l'ensemble de l'organisation. Un projet pilote révèle où se situent les intégrations et met en évidence les lacunes en matière de formation avant qu'elles n'affectent les équipes plus importantes.
Étape 2 : aborder l'intégration dès le début.
Les systèmes mal connectés ajoutent du travail au lieu de le supprimer. Reliez la plateforme SPL aux systèmes d'information réglementaires existants dès le départ pour éviter les saisies de données dupliquées et les mises à jour manquées.
Étape 3 : Intégrez la validation au plan.
Les équipes sous-estiment souvent la durée des activités de qualification. Le logiciel SPL doit être validé dans le cadre de votre système qualité, alors incluez cet effort dans le calendrier plutôt que d'attendre sa mise en service. Coordonner avec l'informatique et l'assurance qualité pour terminer la validation du système dans le cadre des procédures de qualité internes.
Étape 4 : Traitez l'entraînement comme un processus graduel.
L'utilisation pratique est le meilleur professeur. Combinez des modules à votre rythme avec des sessions en direct afin que les professionnels de la réglementation puissent pratiquer les flux de travail de soumission sans perturber les projets en cours. Laissez le temps aux utilisateurs de se familiariser avec les étapes de validation et de dépannage avant le premier cycle de production.
Étape 5 : Exécutez des tests en parallèle.
Exécutez les processus existants et les nouveaux processus côte à côte jusqu'à ce que les résultats soient identiques. Cela permet de détecter les problèmes à un stade précoce tout en préservant la continuité des activités. Prévoyez environ six mois pour les activités de validation et de gestion du changement.
Les équipes qui réduisent les délais pour respecter des délais arbitraires finissent par reconstruire l'intégralité de leur documentation de validation à l'arrivée des auditeurs. Investir correctement ces six mois jette les bases nécessaires pour des soumissions plus rapides et plus précises et moins de stress à long terme.
Critères d'évaluation du logiciel SPL
Le choix du mauvais logiciel SPL peut créer des problèmes de conformité et d'efficacité à long terme. Les opérations réglementaires s'appuient sur des systèmes qui restent stables pendant les mises à jour de la FDA et les longues périodes de soumission, et qui peuvent produire une documentation prête à être auditée à tout moment. Lors de l'évaluation des plateformes, prêtez une attention particulière aux principes fondamentaux qui soutiennent les performances à long terme :
- Assistance à la validation et documentation
- Les fournisseurs doivent fournir des protocoles de validation et des scripts de test conformes aux attentes de la FDA. Sans eux, votre équipe devra faire face à des mois de travail supplémentaire.
- Recherchez des fournisseurs proposant des packages de validation complets comprenant des matrices de traçabilité et des scripts de test exécutés.
- Réactivité à l'évolution de la réglementation
- Lorsque la FDA publie de nouvelles versions de schéma ou de nouvelles spécifications techniques, à quelle vitesse le logiciel reflète-t-il ces modifications ?
- Les fournisseurs performants gardent une longueur d'avance sur les changements de la FDA et vous avertissent avant que les exigences ne changent
- Antécédents en matière de soumission réussie
- Demandez aux fournisseurs quels sont leurs taux d'approbation des demandes pour la première fois
- Les fournisseurs qui ont confiance dans leur logique de validation suivent ces indicateurs
- Des drapeaux rouges qui signalent des problèmes
- Des réponses vagues concernant la conformité à la FDA ou la résistance aux périodes d'essai
- Incapacité à expliquer en détail le processus de mise à jour de leur schéma
- Un retour sur investissement au-delà des coûts de licence
- Calculez le retour sur la base des gains de temps et de la diminution du nombre de refus, plutôt que sur les seuls frais de logiciel
- Un rejet de candidature qui retarde l'accès au marché de huit semaines coûte bien plus cher que les frais d'abonnement annuels
Lorsque vous comparez des fournisseurs, demandez des références à des organisations dont la taille et le volume de soumissions sont similaires aux vôtres. Faites attention à la façon dont chaque fournisseur décrit ses processus de conformité et de mise à jour, car des réponses vagues indiquent généralement un risque. Évaluez le retour sur investissement en termes de gain de temps et de réduction des rejets, et pas seulement en termes de coûts de licence. Les partenaires les plus solides considèrent votre succès en matière de réglementation comme s'il s'agissait du leur, ce qui contribue à accroître votre avantage à chaque soumission.
Mises à jour réglementaires et maintenance logicielle SPL
La FDA publie périodiquement de nouvelles versions de la norme SPL, parfois avec très peu de préavis avant les délais de conformité. Les fournisseurs fiables suivent ces annonces de près et testent leurs logiciels en fonction des modifications de schéma à venir bien avant leur entrée en vigueur. Ils communiquent également les délais à l'avance afin que les clients aient de l'espace pour planifier. En gardant ainsi une longueur d'avance, vous évitez le brouillage qui se produit lorsqu'une mise à jour tombe juste avant un cycle de soumission. Les processus de mise à jour varient selon le modèle de déploiement, mais les fournisseurs sont responsables de la conformité des systèmes à mesure que les exigences de la FDA évoluent.
Une version qui a été validée il y a quelques mois pourrait échouer aujourd'hui si la FDA a mis à jour ses règles ou ses exigences en matière de schéma. Ces fichiers obsolètes sont rapidement rejetés, et les équipes qui reportent la maintenance découvrent souvent des problèmes de soumission uniquement lorsque les délais sont proches.
Tendances futures en matière de développement de logiciels SPL
L'intelligence artificielle et la collaboration dans le cloud trouvent progressivement leur place dans les travaux réglementaires, notamment en ce qui concerne les soumissions SPL. Les outils d'apprentissage automatique permettent désormais d'extraire des informations du contenu d'étiquetage non structuré et de signaler les problèmes de validation potentiels plus tôt dans le processus. Concrètement, ces systèmes apprennent à reconnaître les modèles de données et les structures de formatage, aidant ainsi les équipes à repérer les incohérences qui pourraient autrement passer inaperçues lors de la révision manuelle. Ces systèmes aident les évaluateurs humains en mettant en évidence les domaines à risque, et non en remplaçant le jugement des experts. Dans le même temps, la co-création en temps réel et les flux de travail d'approbation intégrés permettent aux contributeurs de toutes les régions de travailler en parallèle, réduisant ainsi la coordination manuelle et éliminant les problèmes de contrôle de version qui ralentissaient autrefois les cycles de révision. Ensemble, ces technologies rationalisent la validation et accélèrent les délais de soumission.
L'intégration s'étend également au-delà des frontières nationales. Ce qui a commencé comme une initiative de la FDA est devenu un modèle pour d'autres agences de réglementation, de Santé Canada à l'Agence européenne des médicaments. Les plateformes SPL modernes ont de plus en plus besoin d'un support multiformat et multirégional pour éviter la fragmentation des processus à mesure que les organisations s'étendent à l'échelle mondiale.
Des normes émergentes telles que Identification des médicaments (IDMP) représentent la prochaine étape de la gestion des données réglementaires mondiales. Le cadre va au-delà de la SPL et du SPM pour normaliser la manière dont les informations sur les médicaments sont structurées et échangées entre les régions. Les systèmes SPL flexibles peuvent s'adapter à mesure que ces cadres mûrissent, garantissant ainsi la conformité sans retouches constantes. Ces évolutions vont dans le sens de processus de soumission plus rapides et plus transparents, même si l'alignement mondial est toujours en cours.
Sélection de la solution SPL adaptée à votre organisation
La droite Logiciel SPL doit trouver un équilibre entre les exigences réglementaires et la manière dont vos équipes travaillent réellement. Les grands fabricants de produits pharmaceutiques dont les volumes de soumissions sont élevés ont besoin de plateformes d'entreprise qui s'intègrent à tous les systèmes et évoluent en fonction de la croissance. Les petites organisations, ou celles qui n'ont jamais utilisé les soumissions SPL électroniques, bénéficient souvent de solutions plus ciblées ou basées sur les services. Choisissez en fonction de la direction que prend votre organisation plutôt que de ce qui fonctionne aujourd'hui. Les attentes réglementaires se renforcent au fil du temps à mesure que les portefeuilles de produits s'élargissent, et les processus manuels qui semblaient gérables peuvent rapidement se transformer en goulots d'étranglement.
Lorsque vous évaluez les options, allez au-delà du matériel de vente des fournisseurs. Les groupes de travail sectoriels fournissent des informations pratiques émanant de pairs confrontés à des défis de mise en œuvre similaires. Des consultants réglementaires indépendants peuvent évaluer votre environnement de manière objective, tandis que les références clients révèlent les performances des plateformes sous une pression de soumission réelle.
Le succès de la mise en œuvre dépend d'une planification réaliste et d'un soutien exécutif. Établissez des délais qui tiennent compte des activités de validation et de migration des données qui prennent plus de temps que prévu par les fournisseurs. La plupart des équipes ont besoin d'un peu de temps pour s'adapter aux nouveaux flux de travail de soumission et gérer le dépannage sans ralentir les autres tâches. Une fois cette familiarité acquise, le personnel chargé de la réglementation peut abandonner le formatage XML manuel et se concentrer sur l'amélioration de l'exécution des tâches de conformité. Au fil du temps, le processus s'uniformise, les soumissions étant traitées plus rapidement et les problèmes de rejet ayant tendance à diminuer. Une mise en œuvre précipitée permet rarement de gagner du temps et augmente souvent les coûts par la suite.
Alors que le logiciel SPL garantit que vos soumissions répondent aux exigences de mise en forme de la FDA, les plateformes d'inspection de la qualité telles que Verify de GlobalVision garantissent l'exactitude du contenu de votre étiquette avant qu'elle n'entre dans le processus de soumission. Découvrez comment.
