Une simple virgule mal placée sur une ligne de dosage. Une mise en garde oubliée sur l'emballage. Un code-barres illisible au comptoir de la pharmacie. Dans la plupart des secteurs, ce sont des désagréments. Dans l'industrie pharmaceutique, c'est la différence entre un produit sûr et un rappel mettant en danger la vie des patients.
Le conditionnement pharmaceutique est soumis à des contraintes réglementaires plus strictes que presque toute autre catégorie de produits au monde. Chaque élément, du nom du médicament au braille sur l'emballage, existe parce qu'un organisme de réglementation l'exige et qu'un patient en dépend. Pour les équipes de prépresse, d'impression et de conditionnement, cela signifie que la précision n'est pas un objectif. C'est la base.
Voici ce que vous apprendrez dans ce guide :
- Pourquoi le conditionnement pharmaceutique est soumis à une réglementation unique et ce que cela exige de votre équipe
- Les principales exigences de conformité établies par la FDA, l'EMA et d'autres organismes mondiaux
- Les éléments de contenu obligatoires que chaque étiquette et emballage pharmaceutique doit comporter
- Les normes d'inviolabilité et de sécurité enfant incontournables
- Comment le contrôle qualité en prépresse garantit la conformité avant le passage en impression
- Le coût réel de la non-conformité, et comment rester prêt pour les audits à grande échelle
Pourquoi le conditionnement pharmaceutique est soumis à une réglementation unique
Le conditionnement pharmaceutique n'est pas qu'un simple contenant, c'est un outil de communication médicale réglementé. L'étui, l'étiquette, la notice et l'opercule contiennent des informations essentielles sur lesquelles les cliniciens et les patients s'appuient pour doser correctement, stocker en toute sécurité et éviter tout risque. C'est pourquoi les autorités réglementaires examinent chaque détail.
Trois pressions distinguent le conditionnement pharmaceutique de toute autre catégorie :
- La sécurité des patients est en jeu. Une erreur d'étiquetage peut entraîner un mauvais dosage, l'omission d'une mise en garde ou une interaction médicamenteuse dangereuse.
- Les règles ne cessent d'évoluer. Les obligations de sérialisation, les lois linguistiques régionales et les exigences de mise en garde actualisées changent constamment selon les marchés.
- Le volume est incessant. Un seul médicament peut être expédié dans des dizaines de pays, chacun ayant ses propres règles d'étiquetage, de langue et de formatage.
Pour les opérations à fort volume de références se lançant sur les marchés mondiaux, cela représente un défi de taille : maintenir une précision irréprochable tout en étant assez rapide pour respecter les dates de lancement. Trouvez le juste équilibre pour protéger à la fois les patients et votre rentabilité.
Voilà les enjeux. Examinons maintenant les règles.
Exigences de conformité clés : FDA, EMA et au-delà
Le conditionnement pharmaceutique est soumis à un réseau d'organismes de réglementation, chacun ayant ses propres normes. Bien que les spécificités varient selon les régions, l'attente fondamentale est universelle : l'emballage doit être précis, traçable et vérifiable par rapport à un modèle approuvé.
Voici comment les principaux organismes définissent les exigences :
- FDA (États-Unis) : Impose le Structured Product Labeling (SPL) au format XML, le Drug Supply Chain Security Act (DSCSA) pour la sérialisation, ainsi que des règles strictes sur le contenu et le format en vertu du 21 CFR.
- EMA (Union européenne) : Régit l'étiquetage selon les directives européennes, impose le braille sur les boîtes, exige la directive sur les médicaments falsifiés (FMD) pour la sérialisation et impose un conditionnement multilingue dans tous les États membres.
- MHRA (Royaume-Uni): Applique des règles d'étiquetage et de sérialisation spécifiques au Royaume-Uni après le Brexit, exigeant souvent un conditionnement distinct de celui des marchés de l'UE.
- Autres organismes mondiaux : Santé Canada, TGA (Australie), PMDA (Japon) et d'autres ajoutent chacun leurs propres exigences en matière de langue, de format et de sécurité.
La tendance est claire : chaque nouveau marché pénétré ajoute une couche de règles supplémentaire, et donc un risque d'erreur accru. C'est en assurant le suivi manuel de ces couches pour un portefeuille de références grandissant que les équipes se retrouvent submergées.
La stratégie gagnante consiste à vérifier chaque version par rapport à une norme approuvée avant même qu'elle ne parte à l'impression.
Éléments obligatoires pour l'étiquetage et le conditionnement
Les autorités réglementaires ne laissent aucune place à l'interprétation. Le conditionnement pharmaceutique doit comporter des éléments spécifiques et formatés avec précision ; la moindre erreur sur l'un d'eux constitue un point de défaillance potentiel.
Contenu essentiel que tout emballage pharmaceutique doit inclure
- Nom et dosage du médicament : Les noms commerciaux et génériques exacts, ainsi que le dosage précis.
- Posologie et mode d'administration : Instructions d'utilisation claires, conformes aux normes réglementaires.
- Mises en garde et contre-indications : Avertissements encadrés, mentions d'allergènes et informations de sécurité.
- Codes de sérialisation : Identifiants uniques (tels que les codes Data Matrix 2D) pour la traçabilité conformément aux normes DSCSA et FMD.
- Date de péremption et données de lot : Dates de péremption, numéros de lot et dates de fabrication selon les formats approuvés.
- Identifiants uniques des dispositifs (UDI) : Requis pour les produits combinés et les emballages médicament-dispositif.
- Braille : Obligatoire sur les boîtes dans l'UE pour le nom et le dosage du médicament.
- Symboles et mentions réglementaires : Icônes obligatoires, conditions de stockage et mentions légales.
Pourquoi chaque élément comporte des risques
Chaque élément de cette liste doit correspondre exactement au modèle approuvé. Un chiffre inversé dans un code de lot compromet la traçabilité. Une mention d'allergène manquante devient un risque pour la sécurité. Un code-barres illisible bloque le produit lors de la distribution.
En résumé : il n'y a pas d'élément mineur dans le conditionnement pharmaceutique. Chaque détail est un point de contrôle de conformité, et chacun mérite d'être vérifié avant l'impression.
Normes d'inviolabilité et de sécurité enfant pour les emballages
Au-delà des mentions imprimées, les autorités réglementaires imposent des exigences sur la manière dont l'emballage pharmaceutique protège physiquement le produit et ses utilisateurs. Deux normes se distinguent.
Emballages inviolables
Les dispositifs d'inviolabilité , tels que les bandes de rétraction, les étuis scellés ou les bouchons à rupture, fournissent une preuve visuelle claire si un emballage a été ouvert ou altéré. Aux États-Unis, la FDA impose ces dispositifs pour de nombreux produits en vente libre. L'emballage doit présenter des signes évidents de toute effraction, protégeant ainsi les patients contre la contamination ou toute manipulation du produit.
Emballages à sécurité enfant
Emballages à sécurité enfant (CR) sont conçus pour être difficiles à ouvrir pour les jeunes enfants tout en restant accessibles aux adultes. Aux États-Unis, le Poison Prevention Packaging Act définit ces normes ; des règles similaires s'appliquent dans l'UE et sur d'autres marchés. Les plaquettes thermoformées, les bouchons à pression et rotation, ainsi que les systèmes pelables et compressibles répondent tous à cet objectif.
Pour les équipes de conditionnement, ces normes ajoutent des exigences de conception et de structure qui s'ajoutent aux règles relatives au contenu, et les deux doivent être vérifiées conjointement. Une étiquette conforme sur une structure non conforme ne passera pas l'audit.
Contrôle qualité des maquettes et prépresse pour l'industrie pharmaceutique
Voici une vérité que tout professionnel du prépresse connaît par cœur : la presse reproduit fidèlement tout ce que vous lui envoyez, y compris les erreurs. Si une erreur survit à l'étape du prépresse, elle se retrouve jusqu'en pharmacie, multipliée sur chaque unité produite.
C'est pourquoi le contrôle qualité doit intervenir en amont, dès le prépresse, bien avant la gravure des plaques ou le passage en machine.
Pourquoi la relecture manuelle ne suffit plus
La relecture manuelle est utilisée dans l'industrie pharmaceutique depuis des décennies, mais elle atteint ses limites face aux exigences actuelles :
- La fatigue entraîne des oublis. La fatigue visuelle empêche de détecter les subtiles inversions de caractères, les décalages d'espacement et les variations de couleur.
- Le volume submerge les relecteurs. Le nombre élevé de références et les mises à jour fréquentes des étiquettes dépassent les capacités de vérification manuelle.
- Les barrières linguistiques créent des angles morts. Vérifier visuellement des étiquettes destinées à plusieurs marchés dans des langues inconnues est pratiquement impossible.
- Les résultats restent incohérents. Deux relecteurs peuvent aboutir à deux conclusions différentes sur un même fichier.
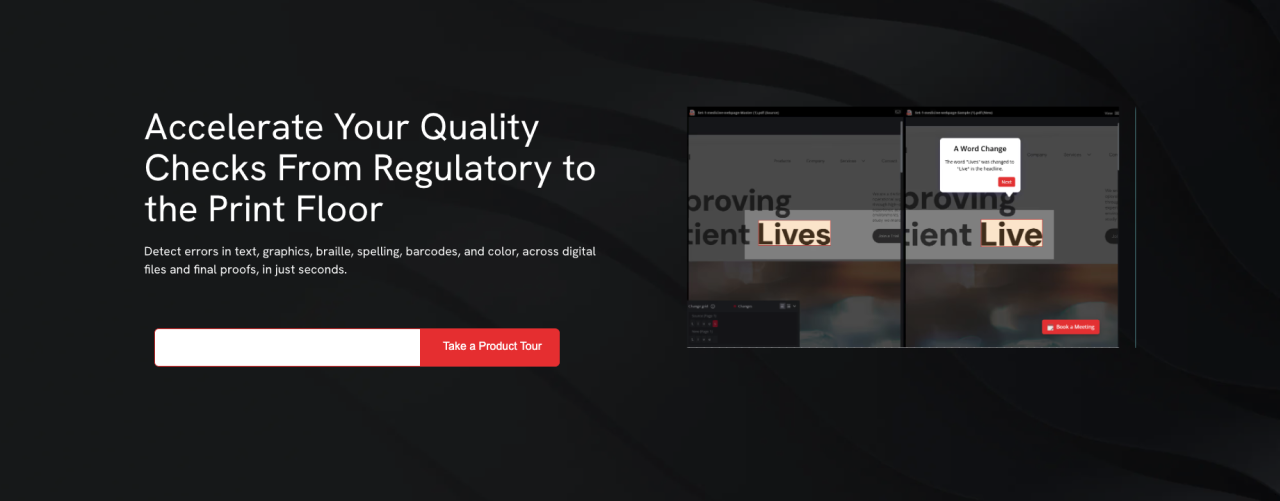
Comment la relecture automatisée garantit la conformité
La relecture automatisée compare chaque fichier à un modèle approuvé et signale les écarts invisibles à l'œil nu. Intégrée au prépresse, elle vérifie la précision avant le lancement de la fabrication des plaques. Les contrôles les plus essentiels incluent :
- La vérification du texte et de l'orthographe pour chaque version linguistique
- La comparaison des graphismes et des visuels par rapport au modèle approuvé
- L'intégrité des codes-barres et des codes de sérialisation pour confirmer que chaque code est scanné et décodé
- Inspection du braille pour la conformité des étuis dans l'UE
- Contrôles des couleurs et du repérage par rapport aux normes de la marque et aux réglementations
En standardisant l'inspection, la relecture automatisée soumet chaque fichier au même contrôle rigoureux et reproductible, sans fatigue, sans approximation et sans goulot d'étranglement. C'est exactement ce qu'exige le conditionnement pharmaceutique.
Le coût de la non-conformité dans le conditionnement pharmaceutique
Il est tentant de considérer une erreur de conditionnement comme un problème mineur. Dans le secteur pharmaceutique, les chiffres racontent une tout autre histoire. Une seule erreur atteignant le marché déclenche une réaction en chaîne affectant la sécurité, les finances et la réputation.
La conclusion est la même pour chaque ligne : une erreur de conditionnement n'est jamais sans conséquence financière. Les erreurs d'étiquetage figurent systématiquement parmi les principales causes de rappels de produits pharmaceutiques, et c'est l'une des plus évitables. Détecter l'erreur lors de la prépresse coûte une fraction du prix d'une détection en rayon.
Bonnes pratiques pour rester prêt pour les audits
La préparation aux audits ne doit pas être une course contre la montre avant l'arrivée de l'inspecteur ; c'est un flux de travail que vous mettez en place une fois et sur lequel vous vous appuyez au quotidien. Voici comment les meilleures équipes de conditionnement pharmaceutique restent prêtes.
1. Établir une source unique de vérité
Verrouillez un fichier maître approuvé pour chaque emballage. Chaque inspection se base sur ce maître, éliminant toute ambiguïté sur ce qui est considéré comme « correct ».
2. Intégrer la relecture automatisée dès le début
Détectez les erreurs en prépresse, pas à l'impression. Lancez des inspections automatisées dès la réception des visuels et après chaque révision. Plus vous détectez une anomalie tôt, plus la correction est rapide et économique.
3. Vérifiez chaque version linguistique et chaque marché
Traitez chaque version régionale comme un fichier distinct nécessitant une vérification complète. La comparaison automatisée de textes rend les contrôles multilingues rapides et fiables, même dans des langues que votre équipe ne maîtrise pas.
4. Intégrez l'inspection aux étapes de validation
Faites de la réussite d'une inspection automatisée une étape obligatoire avant toute transmission de fichier à la photogravure. Le contrôle qualité devient ainsi un verrou incontournable, et non une simple formalité optionnelle.
5. Conservez une piste d'audit complète
Enregistrez chaque inspection, révision et approbation. Lorsque les autorités de régulation se présenteront — et dans le secteur pharmaceutique, elles le feront — vous disposerez de la preuve documentée que chaque emballage était conforme aux spécifications avant son impression.
En combinant ces pratiques, la conformité cesse d'être une source de stress. Elle devient un atout intégré et reproductible qui évolue au même rythme que votre nombre de références et votre portée internationale.
Assurez la conformité constante de vos emballages pharmaceutiques
Dans le conditionnement pharmaceutique, l'« à peu près » n'a pas sa place.
Chaque caractère, code, couleur et élément structurel doit être strictement conforme. Les conséquences d'une erreur sont trop graves pour être laissées au hasard. Les équipes qui réussissent sont celles qui intègrent la précision dans leur flux de travail prépresse, s'appuient sur la relecture automatisée pour éliminer les angles morts humains et considèrent chaque inspection comme un contrôle rigoureux plutôt que comme une simple vérification.
En procédant ainsi, vous atteignez l'équilibre rare exigé par le secteur pharmaceutique : un conditionnement irréprochable, prêt pour l'audit et livré à pleine vitesse sur tous vos marchés.
Questions fréquentes