
Um frasco errôneo desliza por cada checkpoint e chega a uma farmácia hospitalar. As instruções de dosagem parecem rotineiras - um único dígito que ninguém avisa até que seja tarde demais. É um claro lembrete de que a etiqueta farmacêutica compatível permanece uma das salvaguardas mais críticas da indústria farmacêutica.
Introdução à conformidade da etiquetagem farmacêutica
A rotulagem farmacêutica consentânea serve como linha directa entre as empresas farmacêuticas e os profissionais de saúde, fornecendo informações essenciais que afectam directamente a segurança dos doentes. Isto inclui tudo o que é visível nas embalagens e nos contentores, juntamente com os materiais de acompanhamento, assegurar que os profissionais de saúde e os pacientes disponham de informações precisas para tomar decisões de tratamento seguras e informadas.
Os organismos reguladores em todo o mundo mantêm orientações rigorosas, porque a rotulagem de erros pode causar graves prejuízos. Nos Estados Unidos, o Food and Drug Administration (FDA) define e impõe requisitos de rotulagem farmacêutica tanto para medicamentos prescritos como para dispositivos médicos. Em toda a União Europeia, esta função cabe à Agência Europeia de Medicamentos (EMA). Em coordenação com a Saúde no Canadá, Farmacêuticos do Japão e Agência de Dispositivos Médicos (PDMA), e Administração terapêutica de Mercadorias (TGA), estas agências de regulação criam quadros destinados a proteger a saúde pública e a defender a consistência entre os mercados. Os esforços para harmonizar os padrões através do Conselho Internacional para a Harmonização (ICH) continuam, mas o alinhamento total permanece fora de alcance, com cada região a aplicar os seus próprios requisitos regulamentares que as empresas farmacêuticas têm de gerir cuidadosamente. Enquanto isso, a paisagem regulamentar continua a evoluir: a prestação de informação digital está a mudar a forma como os prestadores de cuidados de saúde acessam os dados, e mandatos de serialização estão a redefinir a forma como a rotulagem farmacêutica funciona em todo o sector.
Quadros-chave regulatórios para a etiquetagem farmacêutica
Desde 2006, a Regra de Rotulagem Médica (PLR) definiu o padrão moderno para a rotulagem de medicamentos prescrição médica , trazendo consistência e estrutura a cada produto aprovado. Introduziu uma hierarquia clara para prescrever informação, com uma seção concisa "Destaque" para os profissionais de saúde e regras específicas sobre como o conteúdo remanescente deve aparecer. Estes regulamentos respondem directamente aos erros dos medicamentos que ocorrem quando a informação crítica é enterrada em texto denso. Estes regulamentos rigorosos aplicam-se a todas as categorias de produtos, incluindo os medicamentos contraproducentes a que os consumidores têm acesso sem receitas.
As normas internacionais acrescentam níveis de requisitos para as empresas que operam em todos os mercados. O Resumo de Características da UE (SmPC) e os requisitos de Folha de Dados do Pacote diferem dos formatos da FDA tanto na estrutura como no ênfase do conteúdo. Embora as orientações relativas à Directiva ICH visem harmonizar requisitos técnicos, cada região mantém abordagens distintas. A tabela abaixo mostra como os principais mercados lidam de forma diferente com os requisitos chave de etiquetagem.
Requisitos de rotulagem regulamentares nos principais mercados
| Requisito | FDA (U.S.) | EMA (EU) | Canadá de Saúde |
|---|---|---|---|
| Prescrevendo formato de informação | formato PLR com seção de realces seguido de informações de apresentação completa | SmPC com seções padronizadas | monográfico de produto com três partes: Parte I (profissional de saúde), Parte II (paciente), Parte III (científica) |
| Seções/pedido requerido | Formato estruturado com seções necessárias por diretrizes PLR | Template SmPC padronizado com seções obrigatórias em ordem especificada | Formato estruturado após a orientação de Saúde do Canadá com os elementos necessários de cada parte |
| Atualizar requisitos de notificação | Aprovação prévia das mudanças de eficiência; CBE para atualizações de segurança; Relatório anual para pequenas alterações | Procedimentos de variação com diferentes caminhos de aprovação e cronogramas baseados na significância da mudança | Sistema de notificação diferenciado com base na mudança de significado e nível de risco |
| Padrões de formato eletrônico | SPL (Etiqueta de Produto Estruturada) no formato XML necessário para todas as submissões | Formato eCTD com módulos específicos para rotular conteúdo | Formato eCTD necessário; submissões bilingues em templates padronizados |

O Japão exige que todo o conteúdo da rotulagem seja escrito em japonês e formatado de modo a corresponder a práticas locais de documentação médica. O TGA da Austrália define os seus próprios requisitos para o registo australiano de bens terapêuticos, reflectindo as normas nacionais de clareza e coerência. As empresas que lançam produtos farmacêuticos a nível global devem manter múltiplas versões de rótulo, cada uma das quais cumpre as normas regulamentares locais, garantindo simultaneamente a consistência nas mensagens de segurança essenciais. Gerenciando a versão controle nesses mercados requer sistemas que rastreiam a versão de rótulo onde se aplica fazer com que os fármacos rotulem a conformidade uma disciplina que combina especialistas em assuntos regulatórios com precisões operacionais para navegar no labirinto regulamentar global de hoje.
Componentes essenciais das etiquetas de prescrição de medicamentos
Os rótulos da prescrição de medicamentos fornecem informações diferentes, dependendo de onde elas aparecem. O rótulo imediato do contentor - o que está na garrafa ou no frasco - abrange o básico: o nome da droga, formulário de dosagem, força, rota da administração e muito número. A rotulagem do cartão proporciona mais espaço para trabalhar. O painel de exibição principal mostra o nome e a força estabelecidos juntamente com a quantidade do pacote. Painéis laterais manipulam os destaques da informação prescritiva e detalhes de aconselhamento dos pacientes, além da informação do fabricante. Os regulamentos especificam onde tudo aparece e quão proeminente, até ao tamanho exacto da fonte que torna o produto rotulado como uma disciplina exigente.
Começa por salientar que a superfície onde se prescrevem informações críticas e de segurança passa depois por toda a informação que prescreve. Cada uma das secções segue um formato prescrito, e qualquer desvio requer justificação e aprovação regulamentar. A estrutura mantém dados de segurança essenciais consistentes e fáceis de encontrar, ajudando os revisores e os profissionais de saúde a aceder rapidamente aos detalhes certos. Seções principais incluem:
- Indicações e uso
- Dosagem e administração
- Treinamentos
- Avisos e precauções
- Reações antiverso
- Interações de drogas
- Usar em populações específicas
- Farmacologia Clínica
Seções de informação do paciente precisam atingir um nível de leitura de sexto e oitavo nos EUA, conforme descrito nas diretrizes de legibilidade da FDA. Outras agências de regulação podem aplicar normas de literacia ou formatação diferentes, reflectindo o contexto linguístico e da saúde nas suas regiões. Guias de medicação para medicamentos com riscos graves ajudam os pacientes a entender o seu uso seguro sem os sobrecarregar com dados técnicos. Instruções de uso claras e instruções de dosagem mostram aos pacientes como tomar os seus medicamentos corretamente e o que observar.
Compreensão do Painel de Informação sobre Drogas
O painel de informação sobre medicamentos fornece aos profissionais de saúde os pormenores de que necessitam para prescrever e administrar medicamentos de forma segura. Dentro desse conteúdo, as instruções de armazenamento têm um peso especial porque determinam se um medicamento mantém a sua estabilidade e eficácia ao longo de toda a sua vida de prateleira. Para proteger essa estabilidade, os rótulos definem intervalos de temperatura necessários e explicam como proteger produtos de luz e umidade durante o armazenamento ou transporte. Quando esses limites são ignorados, A degradação pode ocorrer antes de um produto chegar aos pacientes - uma perda evitável de qualidade que realça a necessidade de uma revisão completa das avaliações de qualidade. Alguns medicamentos têm de ser refrigerados, outros têm de ser protegidos da luz, e muitos perdem a sua força se forem expostos à umidade em trânsito. Estes requisitos determinam se os produtos permanecem eficazes ou se transformam em substâncias inúteis (ou nocivas). Quando essas instruções são comunicadas de forma pouco clara ou completamente ignoradas, os produtos perdem a eficácia ou tornam-se potencialmente nocivos.
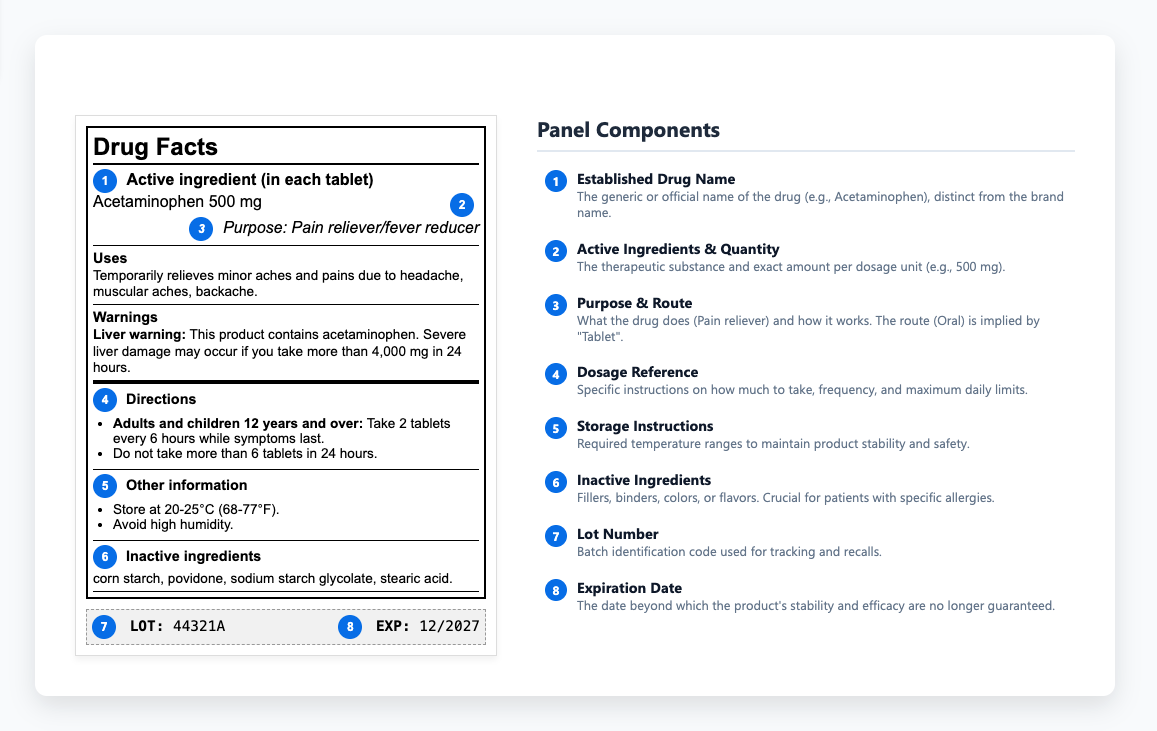
Quando as formulações mudam ou novos dados de segurança aparecem, as vezes as atualizações ficam atrasadas, criando incompatibilidade entre as submissões aprovadas e os rótulos liberados no mercado. Essas incoerências podem inicialmente parecer insignificantes, mas muitas vezes surgem durante as inspecções regulamentares, atrasando as revisões do cumprimento e criando riscos desnecessários. Os auditores comparam o conteúdo do rótulo directamente com as versões aprovadas, para que estas questões raramente permaneçam ocultas durante muito tempo.
Etiquetas de Alerta e Informações de Segurança
Os avisos da caixa negra são a forma mais séria de alerta de segurança da FDA, usado para enfatizar riscos graves ou ameaçadores de vida que devem aparecer em destaque nos rótulos de medicamentos sujeitos a receita médica. Surgem quando as normas de precaução não prevêem danos graves. A FDA acrescenta-as com base em dados pós-mercado que revelam padrões perigosos de reacções adversas, por vezes incluindo mortes. Estes avisos estão no topo da prescrição de informações em fronteiras ousadas, usando uma linguagem directa sobre qual é o risco e quem é mais vulnerável. As treinamentos vão um passo mais longe, identificando quando a droga não deve absolutamente ser usada. Isto inclui interacções perigosas com outros medicamentos, riscos documentados para a gravidez e condições médicas pré-existentes que tornariam o medicamento inseguro.
A informação de reação de anúncios engloba efeitos brandos, esperados até complicações raras, mas graves. A apresentação de dados de frequência, juntamente com esses resultados, ajuda a contextualizar o nível de risco para os pacientes e os revisores. Para reduzir esse risco, as estratégias de mitigação incluem frequentemente o ajustamento de doses para populações específicas, a aplicação de protocolos de monitorização e a orientação dos prestadores de cuidados de saúde sobre aconselhamento e acompanhamento apropriados. Para alguns medicamentos de alto risco, Avaliação de Risco e Estratégias de Mitigação (REMS) mandatam elementos de rótulo adicionais para garantir a segurança do paciente como requisitos de vinculação.

Requisitos de rotulagem especializados para drogas genéricas
O rótulo genérico de medicamentos tem que corresponder ao rótulo do produto de marca quase exatamente, uma exigência que as forças da FDA garantem aos genéricos a mesma segurança e eficácia que o original. Podem divergir em relação a algumas coisas: a informação sobre patentes é deixada de fora por pertencer à marca, enquanto o seu próprio fabricante detalha e códigos NDC substituem o deles. A seção "Como fornecida" também pode refletir suas embalagens específicas.
O trabalho real começa quando o rótulo é alterado. Um novo aviso ou contrainção é adicionado com base em dados pós-mercado e seu rótulo genérico precisa corresponder. O caminho da FDA "Alterações sendo Efeitas" permite que você faça certas atualizações de segurança sem esperar por aprovação, mas continua a ser sua a responsabilidade de apanhar essas mudanças e de as pôr em prática. As empresas que dependem dos reguladores para lhes dizer quando os rótulos estão ultrapassados acabam por ficar para trás, a resolução de problemas após as inspecções regulamentares marca-os em vez de se manterem à frente. Permanecer atual significa acompanhar quando referência os produtos mudam e, em seguida, avaliar como essas atualizações afetam seus produtos específicos. As atualizações precisam divulgar todos os sites de fabricação com documentação que comprove a conformidade durante as auditorias. As pressões geram quando as atualizações de segurança precisam de ação imediata, mas os seus processos normais demoram meses. Os fabricantes de genéricos que lidam com esta boa construção de sistemas de monitorização que capturam mudanças antecipadamente em vez de esperarem pela superfície dos problemas durante a rotulagem de análises de conformidade.
Produtos Biológicos: Considerações específicas de rotulagem
Os produtos biológicos provêm de organismos vivos em vez de síntese química, o que altera o que precisa de figurar nos rótulos. Biologia requer detalhes sobre sua fonte biológica e processos de fabricação juntamente com avisos sobre sistema imunológico que as drogas químicas não desencadeam.
Principais Diferenças de Rotulagem
| Rotulando Elemento | Pequenos Molecules Drugs | Produtos Biológicos |
|---|---|---|
| Detalhes de fabricação | Descrição geral do processo | Fonte biológica detalhada, informação sobre linha celular e controles de processo que afetam as características do produto |
| Requisitos de armazenamento | Frequentemente a temperatura da sala é estável com especificações padrão | Normalmente requer refrigeração com estreitos intervalos de temperatura; muitas proteção de luz e são sensíveis à agitação |
| Informações de imunogenicidade | Não se aplica | Avisos necessários sobre potenciais respostas imunológicas e desenvolvimento de antidopagem |
| Relacionamento biosimilar | Não se aplica | Deve incluir declaração explicando a relação com referência à biológica e natureza de similaridade |
| Vigilância pós-mercado | Relatório padrão adverso de eventos | Aprimorado monitoramento de programas com participação específica de registro ou requisitos de rastreamento de longo prazo |
Os requisitos de armazenamento de biologia vão além das especificações padrão porque esses produtos se degradam facilmente em condições que não afetariam as drogas químicas. A maior necessidade de refrigeração em intervalos apertados de temperatura e de exposição curta ao calor, luz ou agitação pode torná-los ineficazes ou potencialmente nocivos. Os rótulos descrevem como os produtos devem ser armazenados e transportados, detalhando os limites de temperatura e os passos de processamento confirmados durante o desenvolvimento.
Os avisos de imunogenicidade explicam como o sistema imunitário pode reagir a produtos biológicos. Os rótulos explicam o risco de desenvolvimento de antidopagem e resumem a forma como os ensaios clínicos avaliaram esse risco, incluindo o que as conclusões significam para os pacientes. Os prestadores de cuidados de saúde utilizam esta informação para monitorizar a resposta ao tratamento e ajustar a terapia quando se desenvolvem reacções imunitárias. A vigilância pós-mercado estende-se além dos relatos de eventos adversos padrão, com etiquetas especificando requisitos de participação do registro ou monitoramento a longo prazo que rastreia os ensaios clínicos dos efeitos não podem ser totalmente capturados.
Desafios e Soluções de Etiqueta Multilíngue
As empresas farmacêuticas que operam globalmente enfrentam desafios distintos ao criar rótulos que atendam a diversos requisitos linguísticos, mantendo a precisão e a conformidade da regulamentação.
Desafio: qualidade de tradução e precisão cultural
A terminologia médica não traduz diretamente entre línguas e o contexto cultural forma como os pacientes entendem informação sobre saúde. Uma tradução tecnicamente precisa pode falhar completamente a forma como as pessoas num mercado alvo realmente discutem conceitos médicos. Serviços de tradução padrão não possuem as exigências de rotulagem farmacêutica especializada em conhecimento.
Uma tradução eficaz requer especialistas que entendem tanto a terminologia médica como as expectativas reguladoras de cada mercado-alvo. Os tradutores precisam de fluência nativa e conhecimento funcional do domínio farmacêutico para garantir a precisão e o contexto. A FDA e outras agências reguladoras descrevem quando os materiais traduzidos precisam de aprovação formal ou de um tradutor certificado, com regras específicas variando por região.
Desafio: Realizando diversos requisitos de idioma do mercado
Muitos mercados encarregam várias línguas num único rótulo. O Canadá requer inglês bilíngue e francês. A Suíça precisa de alemão, francês e italiano. A Bélgica exige francês, holandês e alemão. Preencher múltiplas traduções completas em etiquetas com restrições de espaço, força compromissos difíceis entre legibilidade e cumprimento dos requisitos regulamentares. As empresas abordam isso trabalhando com designers de etiquetas especializados em embalagens farmacêuticas multilíngues, usar técnicas como dobras de acordião, rótulos de livros ou hierarquias de informações diferenciadas que priorizam informações de segurança críticas.
Desafio: Coordenar atualizações entre idiomas e mercados
Quando a informação de segurança muda no rótulo principal, cada versão traduzida precisa de ser atualizada. Prazos regulamentares nos mercados raramente se alinham, criando períodos em que diferentes países apresentam nos rótulos informações de segurança diferentes.
A tecnologia oferece soluções aqui. Os sistemas de memória de tradução mantêm a consistência terminológica entre os produtos, enquanto as bases de dados de terminologia garantem termos técnicos de tradução uniformemente toda vez que aparecem. Os sistemas de gestão de fluxo de trabalho juntam isso rastreando quais versões de rótulo existem em quais idiomas. quais mercados aprovaram cada versão e onde são necessárias atualizações. Esses sistemas ajudam as empresas a manter o cumprimento, assinalando lacunas e coordenando o controlo da versão em todas as operações globais antes de surgirem problemas com inspecções regulatórias.
Serialização e Faixa & Requisitos de Rastreamento
Os regulamentos de serialização mudaram a forma como a etiquetagem farmacêutica funciona, exigindo que cada pacote carrege um identificador único que o rastreie através da cadeia de fornecimento. Isto aborda problemas reais, incluindo a contrafacção e a necessidade de executar operações de recolha rápida quando surgem questões de segurança. Actualmente, em todo o mundo, a serialização dos mandatos, embora os prazos de execução tenham variado de mercado. A Lei dos Estados Unidos sobre a Segurança da Cadeia de Droga e da Toxicodependência exigiu o seu cumprimento até 2023, enquanto a Directiva relativa aos Medicamentos Falsificados da UE entrou em vigor em 2019. Outros mercados seguiram os seus próprios horários, criando uma implantação global escalonada que exigia que as empresas gerenciassem diferentes requisitos simultaneamente. Estes regulamentos requerem pacotes para transportar identificadores de produtos com vários elementos de dados - Códigos Nacionais de Droga e números de série únicos, juntamente com muitos números e datas de expiração - todos em formatos tanto humanos como máquinas podem ler.
A tecnologia depende principalmente dos códigos de barras da Matriz de Dados 2D, que armazenam grandes quantidades de informação em um formato compacto. Estes códigos aparecem em todas as fases da produção, desde linhas de fabrico e sistemas de controlo de qualidade até bases de dados de distribuição que rastreiam cada identificador único. Para a maioria das equipas, a verdadeira complexidade surge quando estes requisitos têm de coexistir com rótulos já cheios de conteúdo obrigatório. Códigos de barras não podem obscurecer o texto necessário, e os números de série devem aparecer em formatos legíveis para humanos e para máquinas. Ao mesmo tempo, é necessário que os dados de serialização permaneçam nas suas posições tradicionais, de modo a respeitarem processos de rotulagem farmacêutica de longa data. Na prática, as empresas estão agora a conceber rótulos que satisfazem duas gerações de expectativas regulamentares, mantendo ao mesmo tempo clareza e legibilidade.
Controle de qualidade na etiquetagem Farmacêutica
Avaliações de design marcam a comparação de etiquetas propostas contra submissões reguladoras aprovadas. O texto precisa exatamente corresponder os gráficos devem aparecer como aprovado, a formatação deve seguir as especificações. Mesmo os desvios menores requerem documentação e muitas vezes desencadeiam notificações regulamentares, razão pela qual essas revisões estabelecem a linha base para toda a verificação subsequente. Uma vez que as provas do fornecedor chegam, as ferramentas de inspecção fortalecidas por IA ajudam a receber equipes de garantia de qualidade verificar se os arquivos correspondem a documentos mestres aprovados antes de a impressão começar:
- Comparação de Artwork verifica provas de mestres aprovados para pegar deslocamentos de layout ou elementos que faltam.
- Código de barras e verificação do código QR confirma que os dados codificados estão corretos e que cada código segue a formatação necessária antes de imprimir
- Reconhecimento de caracteres óptico (OCR) identifica diferenças de texto entre provas e arquivos mestres aprovados para garantir que nenhuma alteração não intencional foi introduzida durante as atualizações
- UDetecção de mudança intendida verifica se correções anotadas de versões anteriores de arquivo foram aplicadas corretamente, ajudando as equipes a confirmar que as atualizações solicitadas foram implementadas antes da produção.
Esses sistemas funcionam continuamente durante as execuções de produção, analisar arte e texto para sinalizar problemas imediatamente em vez de descobri-los depois de milhares de unidades terem sido impressas A supervisão humana lida com excepções e fornece a aprovação final, mas a inspecção automatizada transporta a maior parte do trabalho de verificação em escala.
A documentação une tudo, provando aos reguladores que os procedimentos foram efectivamente seguidos. Registros em lote capturam o que acontece durante a produção - que inspecções, andam, quais resultados voltaram, como desvios foram resolvidos, quem aprovou a libertação. Durante as inspecções regulamentares, estes registos demonstram que o seu sistema de controlo de qualidade funciona efectivamente e não apenas como procedimentos escritos. A documentação em falta cria preocupações de conformidade, mesmo quando as próprias etiquetas estão corretas, porque os reguladores não podem verificar o processo funcionou como projetado. As empresas que integram estas medidas de controlo de qualidade desde a concepção até à documentação pegam erros de rotulagem antes de se tornarem problemas de mercado. enquanto aqueles que dependem de verificações reativas descobrem problemas durante as auditorias quando as correções são muito mais caras.
Ferramentas de automação para a etiqueta de conformidade
Estas plataformas gerenciam fluxos de trabalho onde várias equipes revisam etiquetas antes da produção, garantindo rótulos compatíveis em cada etapa da revisão. O conteúdo se move da pesquisa e desenvolvimento de produtos através de revisão regulatória, em seguida, na fase de design e marketing para layout, verificação de marcas e reivindicações antes de atingir a garantia de qualidade para verificações de precisão finais. O sistema rastreia quem reviu o quê e quando, criando a trilha de auditoria exige inspecções regulamentares. As empresas podem dimensionar estes fluxos de trabalho para além das cadeias de e-mail e unidades compartilhadas que gerenciam.
A validação segue o mesmo rigor exigido para qualquer sistema farmacêutico de fabrico. O processo começa com a qualificação da instalação para confirmar a configuração correta, move através da qualificação operacional que testa todas as funções, e conclui com a qualificação de desempenho demonstrando precisão consistente durante as operações normais. A validação prova que o sistema automatizado previne confiavelmente erros de etiquetagem, em vez de introduzir novos.
A integração com sistemas reguladores de gestão de informação liga o conteúdo aprovado diretamente à produção de rótulos. Atualizações de prescrever informações depois que as aprovações da FDA fluem diretamente para rótulos relevantes sem que ninguém copie manualmente textos entre os sistemas, que corta erros de transcrição enquanto acelera ciclos de mudança. Os principais benefícios incluem atualizações mais rápidas do rótulo e um controle de versão mais apertado em todos os sites globais, suportado por fluxos de trabalho de revisão estruturados que reforçam a conformidade global da regulação. As empresas vêem ganhos de eficiência operacional, reduzindo os pontos de contacto manuais e fazendo com que a visibilidade em tempo real passe a marcar o estado das operações de fabrico.
Melhores Práticas de Submissões Regulatórias
Envolver as questões regulamentares durante o desenvolvimento do produto, por isso considerações de rotulagem podem moldar como você planeja os ensaios clínicos e coletar dados. Esperar até o tempo de arquivamento significa que você está travado tentando fazer com que seus dados suportem a rotulagem alegações que ele não foi projetado para provar.
A sua submissão precisa de etiquetagem no formato que a agência espera. O FDA quer o formato estruturado de etiquetagem de produtos (SPL), um padrão XML que seus sistemas podem processar electronicamente. O conteúdo segue as normas da indústria - as secções aparecem em ordem prescrita, incluindo todos os elementos necessários. Acerte as especificações técnicas e a sua submissão é rejeitada por causa da formatação, ao invés de quaisquer problemas reais de segurança ou eficiência.
Rastreando o que mudou da versão atualmente aprovada, separa as atualizações de rotulagem suaves de caixas de cabeçalhos regulatórias. Algumas alterações precisam de aprovação prévia antes da implementação, outras têm de passar por disposições "alterações em vigor" e outras têm de ser relatadas anualmente. Misture estes problemas e você está criando problemas de conformidade que dificultam a obtenção de atualizações no mercado. Submissões eletrônicas substituíram o papel, com o conteúdo estruturado do Processamento do Gateway da FDA diferente dos PDFs. Muitas empresas introduzem serviços especializados para lidar com os requisitos técnicos e garantir a precisão. Após a aprovação, os seus sistemas precisam garantir que apenas versões aprovadas cheguem à produção. Altere o controle que realmente funciona impede que rótulos antigos deslizar após a aprovação de novas versões.
Requisitos para treinamento e documentação
Todos os envolvidos em operações de rotulagem precisam entender o que estão a fazer e por que isso importa, e você precisa provar isso. Essas sessões não são eventos únicos. A formação contínua mantém as equipas a par de mudanças regulamentares e de novos sistemas. A verdadeira medida não é se a formação acontece, mas se as equipas compreendem os requisitos actuais quando os inspectores chegam.
Procedimentos operacionais normais (SOPs) documento como sua empresa lida com cada passo do processo de rotulagem, mostrando aos reguladores que seguem um processo definido, em vez de tomar decisões de forma diferente, dependendo de quem trabalha nesse dia. O SOPs precisa cobrir:
- Como os arquivos artísticos são gerenciados e a versão é controlada
- Como as aprovações fluem pela organização
- Como as mudanças são iniciadas, revisadas e implementadas
- Como os desvios e problemas são investigados e resolvidos
Escrever SOPs é simples: segui-los de forma consistente e actualizá-los quando as práticas de rotulagem mudam é onde as empresas se debatem. Os registros demonstram que o seu sistema de qualidade funciona como projetado, não apenas no papel. É preciso que demonstrem que o pessoal qualificado seguiu os procedimentos aprovados, as inspecções foram concluídas conforme planeado, e quaisquer desvios foram devidamente investigados. Durante as inspecções regulamentares, estes ficheiros respondem à pergunta mais básica: fez de facto o que os seus procedimentos dizem que faz? O treinamento estende-se para além da etiquetagem de especialistas a qualquer pessoa que toque no processo, desde funcionários dos assuntos regulamentares escrevendo conteúdo a designers gráficos que criam obras de arte até pessoal de garantia de qualidade que aprova corridas de produção. Equipes que enfatizam programas de treinamento fortes tendem a ter menos problemas de conformidade durante as auditorias e podem responder mais rapidamente quando os requisitos mudam. A alternativa é descobrir lacunas quando os reguladores as encontram, o que custa muito mais a corrigir.
Tendências futuras na etiquetagem farmacêutica
A Etiqueta digital está indo além das versões PDF de inserções de papel. Códigos QR no link de pacotes agora diretamente para prescrever informações que atualizam em tempo real permitir que os profissionais de saúde tenham acesso aos dados de segurança atuais, em vez de confiar em inserções impressas que possam ter meses de idade. A FDA reconhece que os rótulos em papel têm limites, em especial para produtos complexos em que uma informação abrangente excede restrições espaciais razoáveis, mas os reguladores não abandonaram a rotulagem física. A informação principal de segurança ainda requer presença em embalagens para garantir o acesso sem dispositivos digitais.
A pressão ambiental está a reformular as decisões sobre embalagens de uma forma que, por vezes, está em conflito com as exigências regulamentares. O sector farmacêutico enfrenta a exigência de reduzir os resíduos e de utilizar materiais sustentáveis, objectivos que podem colidir com regulamentos que especificam determinadas construções de embalagens. A União Europeia tem impulsionado requisitos particularmente agressivos em matéria de embalagens sustentáveis que afectam a concepção e a produção.
Etiquetas inteligentes com chips NFC ou tecnologias incorporadas verificam a autenticidade do produto durante o rastreamento do uso e monitoram a exposição da temperatura durante a distribuição. Essas capacidades levantam novas questões regulamentares sobre privacidade e segurança de dados, bem como a extensão da funcionalidade de rótulos inteligentes em território de dispositivos médicos. A tecnologia existe; o quadro regulamentar continua a constituir.
Recursos para profissionais de etiquetagem farmacêutica
Associações profissionais, como a Sociedade de Assuntos Regulatórios (RAPS) e a Associação de Informação sobre Drogas (DIA) conectam você com pessoas que enfrentam desafios de rotulagem semelhantes por meio de conferências e grupos de trabalho. Você percebe como os reguladores se aproximam de novos requisitos enquanto vêem como as outras empresas resolvem os problemas com que você está lidando. O RAPS também oferece uma certificação de assuntos reguladores que cobre competências de rotulagem, enquanto programas de treinamento especializados abordam lacunas específicas quando as equipes precisam obter novos regulamentos ou tecnologias.
O apoio externo vem de duas direcções. As empresas de consultoria trazem experiência regulatória para navegar em submissões complexas, particularmente quando se trata de requisitos desconhecidos ou prazos apertados. Provedores de tecnologia oferecem plataformas que lidam com o gerenciamento de versão entre mercados enquanto coordenam alterações e aprovações. A escolha entre eles muitas vezes depende de saber se o seu desafio é compreender o que os reguladores querem ou gerenciar a complexidade operacional de fazer isso.
Para equipes que gerenciam conteúdos de alta participação, a precisão não é opcional. GlobalVison Verify dá às equipes a confiança de que cada versão aprovada está pronta para inspeção.
