
Un flaconcino erroneamente etichettato scivola attraverso ogni punto di controllo e raggiunge una farmacia ospedaliera. Le istruzioni per il dosaggio sembrano routine - una singola cifra fuori che nessuno nota fino a quando non è troppo tardi. È un forte promemoria che l'etichettatura farmaceutica conforme rimane una delle salvaguardie più critiche nell'industria farmaceutica.
Introduzione alla conformità farmaceutica all'etichettatura
L'etichettatura farmaceutica conforme funge da linea diretta tra le aziende farmaceutiche e gli operatori sanitari, fornendo informazioni essenziali che influiscono direttamente sulla sicurezza dei pazienti. Questo include tutto ciò che è visibile sull'imballaggio della droga e sui contenitori insieme ai materiali di accompagnamento, garantire che i medici e i pazienti dispongano di informazioni accurate per prendere decisioni di trattamento sicure e informate.
Gli organismi di regolamentazione in tutto il mondo mantengono rigide linee guida perché gli errori di etichettatura possono causare gravi danni. Negli Stati Uniti, la Food and Drug Administration (FDA) definisce e impone requisiti di etichettatura farmaceutica sia per i farmaci da prescrizione che per i dispositivi medici. In tutta l'Unione europea, questo ruolo ricade sull'Agenzia europea per i medicinali (EMA). In coordinamento con Health Canada, Japanese Pharmaceuticals and Medical Devices Agency (PDMA), e Therapeutic Goods Administration (TGA), queste agenzie di regolamentazione creano quadri destinati a proteggere la salute pubblica e sostenere la coerenza tra i mercati. Efforts to harmonize standards through the International Council for Harmonisation (ICH) continue, but full alignment remains out of reach, with each region applying its own regulatory requirements that pharmaceutical companies must manage carefully. Nel frattempo, il panorama normativo continua ad evolversi: la fornitura di informazioni digitali sta cambiando il modo in cui i fornitori di assistenza sanitaria accedono ai dati, e mandati di serializzazione stanno ridefinendo come funzionano i processi di etichettatura farmaceutica in tutto il settore.
Principali quadri normativi per l'etichettatura farmaceutica
Dal 2006, la Regola Medico di Etichettatura (PLR) della FDA ha definito lo standard moderno per l'etichettatura farmaco di prescrizione , portare consistenza e struttura a ogni prodotto approvato. Ha introdotto una chiara gerarchia per la prescrizione di informazioni, con una sezione “Highlight” concisa per gli operatori sanitari e regole specifiche su come deve apparire il contenuto rimanente. Questi regolamenti rispondono direttamente agli errori di medicazione che si verificano quando le informazioni critiche vengono sepolte in un testo denso. Questi severi regolamenti si applicano a tutte le categorie di prodotti, tra cui i farmaci contro cui i consumatori accedono senza prescrizioni.
Le norme internazionali aggiungono livelli di requisiti per le imprese che operano su tutti i mercati. The EU's Summary of Product Characteristics (SmPC) and Package Leaflet requirements differ from FDA formats in both structure and content emphasis. Mentre le linee guida ICH mirano ad armonizzare i requisiti tecnici, ogni regione mantiene approcci distinti. La tabella seguente mostra come i principali mercati gestiscono i requisiti di etichettatura dei tasti in modo diverso.
Requisiti in materia di etichettatura per i principali mercati
| Requisito | FDA (U.S.) | EMA (EU) | Salute Canada |
|---|---|---|---|
| Formato delle informazioni di prescrizione | Formato PLR con sezione Highlights seguita da Informazioni complete di prescrizione | RCP con sezioni standardizzate | Monografia del prodotto con tre parti: Parte I (operatore sanitario), Parte II (paziente), Parte III (scientifico) |
| Sezioni richieste/ordine | Formato strutturato con sezioni richieste per linee guida PLR | Modello di RCP standardizzato con sezioni obbligatorie nell'ordine specificato | Formato strutturato in base alla guida di Health Canada con gli elementi richiesti in ogni parte |
| Aggiorna i requisiti di notifica | Approvazione preventiva per le modifiche di efficacia; CBE per gli aggiornamenti di sicurezza; segnalazione annuale per modifiche minori | Procedure di variazione con percorsi di approvazione e temporali diversi in base al significato delle variazioni | Sistema di notifica a livelli basato sul cambiamento di significatività e livello di rischio |
| Norme in formato elettronico | SPL (Structured Product Labeling) in formato XML richiesto per tutte le presentazioni | formato eCTD con moduli specifici per il contenuto dell'etichettatura | formato eCTD richiesto; contributi bilingui in modelli standardizzati |

Il Giappone incarica che tutti i contenuti di etichettatura siano scritti in giapponese e formattati per corrispondere alle pratiche locali di documentazione medica. Il TGA australiano definisce i propri requisiti per il Registro australiano dei Beni Terapeutici, riflettendo gli standard nazionali per chiarezza e coerenza. Le aziende che lanciano prodotti farmaceutici in tutto il mondo devono mantenere diverse versioni di etichette, ciascuna conforme agli standard normativi locali, garantendo al contempo la coerenza dei messaggi di sicurezza di base. Gestire il controllo di versione in questi mercati richiede che i sistemi che tracciano quale versione di etichetta si applica dove, rendere l'etichettatura pharma conformità una disciplina che combina competenze in materia di regolamentazione con precisioni operative per navigare il labirinto normativo globale di oggi.
Componenti essenziali delle etichette di farmaci di prescrizione conformi
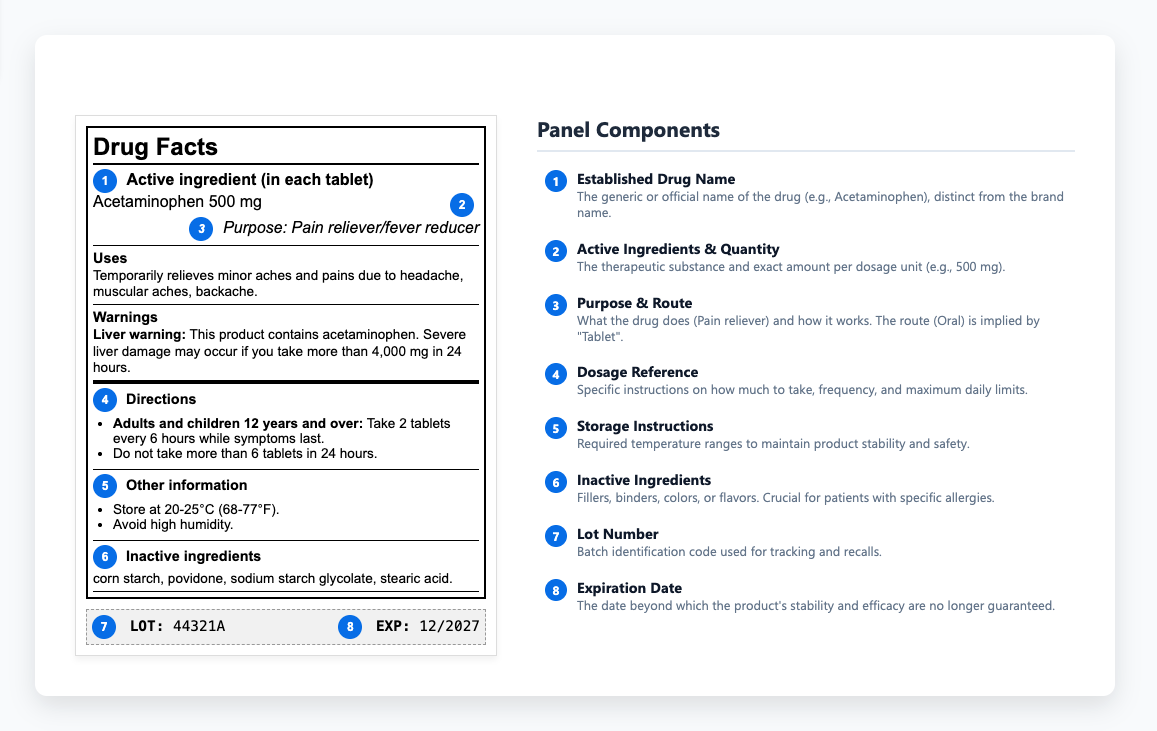
Le etichette di prescrizione dei farmaci forniscono informazioni diverse a seconda di dove appaiono. L'etichetta del contenitore - che cosa c'è sul flacone o sul flaconcino - copre le basi: il nome del farmaco, forma di dosaggio, forza, via di somministrazione e numero del lotto. Etichettatura del cartone offre più spazio per lavorare. Il pannello principale mostra il nome e la forza stabiliti insieme alla quantità del pacchetto. I pannelli laterali gestiscono le informazioni di prescrizione e i dettagli di consulenza del paziente, oltre alle informazioni del produttore. I regolamenti specificano dove appare tutto e come prominente, giù alle dimensioni esatte dei caratteri che rendono l'etichettatura del prodotto una disciplina esigente.
Si inizia con evidenzia che la prescrizione critica della superficie e le informazioni sulla sicurezza, quindi passa attraverso le informazioni complete di prescrizione. Ogni sezione segue un formato prescritto e ogni deviazione richiede giustificazione e approvazione regolamentare. La struttura mantiene i dati essenziali di sicurezza coerenti e facili da trovare, aiutando i revisori e gli operatori sanitari ad accedere rapidamente ai dettagli giusti. Le sezioni principali comprendono:
- Indicazioni e utilizzo
- Dosaggio e somministrazione
- Controindicazioni
- Avvertenze e precauzioni
- Reazioni avverse
- Interazioni farmacologiche
- Uso in popolazioni specifiche
- Farmacologia clinica
Le sezioni di informazione per i pazienti devono raggiungere un livello di lettura da sesto a ottavo grado negli Stati Uniti, come indicato nelle linee guida sulla leggibilità dell’FDA. Altre agenzie di regolamentazione possono applicare diversi standard di alfabetizzazione o formattazione, che riflettono il contesto linguistico e sanitario nelle loro regioni. Le guide mediche per farmaci con gravi rischi aiutano i pazienti a capire l'uso sicuro senza travolgerli di dettagli tecnici. Istruzioni chiare per l'uso e istruzioni per il dosaggio mostrano ai pazienti come assumere i loro farmaci correttamente e cosa guardare.
Comprendere il gruppo di informazioni sulla droga
Il pannello informativo del farmaco fornisce agli operatori sanitari i dettagli necessari per prescrivere e somministrare i farmaci in modo sicuro. All'interno di quel contenuto, le istruzioni di conservazione hanno un peso speciale perché determinano se un farmaco mantiene la sua stabilità ed efficacia per tutta la sua durata di conservazione. Per proteggere tale stabilità, le etichette definiscono le gamme di temperatura richieste e spiegano come proteggere i prodotti dalla luce e dall'umidità durante la conservazione o il trasporto. Quando questi limiti sono trascurati, la degradazione può verificarsi prima che un prodotto raggiunga mai i pazienti - una perdita evitabile di qualità che sottolinea la necessità di una revisione approfondita del QA. Alcuni farmaci devono essere refrigerati, altri protetti dalla luce e molti perdono potenza se esposti all'umidità durante il transito. Questi requisiti determinano se i prodotti rimangono efficaci o si trasformano in sostanze inutili (o nocive). Quando queste istruzioni vengono comunicate in modo non chiaro o completamente ignorate, i prodotti perdono efficacia o diventano potenzialmente dannosi.
Quando le formulazioni cambiano o compaiono nuovi dati di sicurezza, gli aggiornamenti delle etichette a volte sono in ritardo, creando incongruenze tra le presentazioni approvate e le etichette rilasciate sul mercato. Tali incongruenze possono sembrare lievi in un primo momento, ma spesso si presentano durante le ispezioni normative, rallentando le revisioni di conformità e aggiungendo rischi inutili. I revisori dei conti confrontano il contenuto dell'etichetta direttamente con le versioni approvate, quindi questi problemi raramente rimangono nascosti per molto tempo.
Etichette di avvertimento e informazioni di sicurezza
Gli avvertimenti della scatola nera sono la forma più grave di allarme di sicurezza della FDA, usato per sottolineare rischi pericolosi per la vita o gravi che devono apparire in primo piano su etichette di farmaci da prescrizione. Si manifestano quando le precauzioni standard non riescono a prevenire danni gravi. La FDA li aggiunge sulla base di dati post-mercato rivelando modelli di reazioni avverse pericolose, a volte compresi i decessi. Questi avvertimenti sedersi in cima alle informazioni di prescrizione in grassetto frontiera, utilizzando il linguaggio diretto su ciò che il rischio è e chi è più vulnerabile. Le controindicazioni vanno un ulteriore passo avanti identificando quando il farmaco assolutamente non dovrebbe essere usato. Ciò include interazioni pericolose con altri farmaci, rischi di gravidanza documentati e condizioni mediche preesistenti che renderebbero il farmaco pericoloso.
Le informazioni sulla reazione avversa coprono effetti lievi, attesi attraverso complicazioni rare ma gravi. La presentazione dei dati sulla frequenza insieme a questi risultati aiuta a mettere il livello di rischio nel contesto per i pazienti e gli esaminatori. Per ridurre tale rischio, le strategie di mitigazione spesso includono l'aggiustamento delle dosi per popolazioni specifiche, l'applicazione di protocolli di monitoraggio e la guida dei fornitori di assistenza sanitaria su consulenze e follow-up adeguati. Per alcuni farmaci ad alto rischio, i programmi Risk Evaluation and Mitigation Strategies (REMS) richiedono ulteriori elementi di etichettatura per garantire la sicurezza del paziente come requisiti vincolanti.

Requisiti speciali di etichettatura per farmaci generici
L'etichettatura generica dei farmaci deve corrispondere quasi esattamente all'etichetta del prodotto di marca, un requisito che la FDA si impegna a garantire che i generici forniscano la stessa sicurezza ed efficacia dell'originale. Si può differire su alcune cose: le informazioni di brevetto viene lasciato fuori dal momento che appartiene al marchio, mentre i dettagli del produttore e i codici NDC sostituiscono i loro. La sezione "Come Fornito" può anche riflettere il vostro imballaggio specifico.
Il vero lavoro inizia quando cambia l'etichetta di marca. Un nuovo avvertimento o controindicazione viene aggiunto in base ai dati post-marketing, e la tua etichetta generica deve corrispondere. Il percorso "Cambiamenti Effettivi" della FDA ti permette di effettuare alcuni aggiornamenti di sicurezza senza attendere l'approvazione, ma voi avete ancora la responsabilità di cogliere questi cambiamenti e di agire su di essi. Le aziende che si affidano alle autorità di regolamentazione per dire loro quando le etichette sono superate finiscono indietro, risolvere i problemi dopo le ispezioni regolamentari li bandierano invece di restare avanti. Rimanere corrente significa tracciare quando i prodotti di riferimento cambiano, quindi valutare come tali aggiornamenti influenzano i prodotti specifici. Gli aggiornamenti devono essere pubblicati in tutti i siti di produzione con documentazione che dimostri la conformità durante gli audit. La pressione si accumula quando gli aggiornamenti di sicurezza richiedono un'azione immediata, ma i processi normali richiedono mesi. Generici produttori che gestiscono questo pozzo costruire sistemi di monitoraggio che catturano i cambiamenti precocemente piuttosto che aspettare i problemi di superficie durante le revisioni di conformità dell'etichettatura.
Prodotti Biologici: Considerazioni Specifiche Sull'Etichettatura
I prodotti biologici provengono da organismi viventi invece della sintesi chimica, che cambia ciò che deve apparire sulle etichette. La biologia richiede dettagli sulla loro fonte biologica e i processi di produzione insieme a avvertimenti sulle risposte del sistema immunitario che i farmaci chimici non innescano.
Differenze Di Etichettatura Chiave
| Elemento Etichettatura | Piccole Farmaci Molecolari | Prodotti Biologici |
|---|---|---|
| Dettagli di fabbricazione | Descrizione generale del processo | Fonte biologica dettagliata, informazioni sulla linea cellulare e controlli di processo che influiscono sulle caratteristiche del prodotto |
| Requisiti di stoccaggio | Spesso la temperatura ambiente stabile con specifiche standard | In genere richiedono refrigerazione con intervalli di temperatura ristretti; molti hanno bisogno di protezione della luce e sono sensibili all'agitazione |
| Informazioni sull'immunogenicità | Non applicabile | Avvertenze richieste sulle potenziali risposte immunitarie e sullo sviluppo di anticorpi anti-farmaco |
| Relazione biosimilare | Non applicabile | Deve includere la dichiarazione che spiega il rapporto con riferimento biologico e la natura della somiglianza |
| Sorveglianza post-mercato | Segnalazione di eventi avversi standard | Programmi di monitoraggio potenziati con specifici requisiti di partecipazione del registro o di tracciamento a lungo termine |
I requisiti di stoccaggio per la biologia vanno al di là delle specifiche standard perché questi prodotti si degradano facilmente in condizioni che non influirebbero sui farmaci chimici. La maggior parte delle necessità di refrigerazione entro intervalli di temperatura ristretti, e breve esposizione a calore, luce o agitazione può renderli inefficaci o potenzialmente dannosi. Le etichette delineano le modalità di conservazione e trasporto dei prodotti, precisando i limiti di temperatura e le fasi di manipolazione confermate durante lo sviluppo.
Le avvertenze di immunogenicità spiegano come il sistema immunitario può reagire ai prodotti biologici. Le etichette spiegano il rischio di sviluppare anticorpi anti-farmaco e sintetizzano il modo in cui gli studi clinici hanno valutato tale rischio, includendo ciò che i risultati significano per i pazienti. Gli operatori sanitari utilizzano queste informazioni per monitorare la risposta al trattamento e regolare la terapia quando si sviluppano reazioni immunitarie. La sorveglianza post-mercato va oltre la segnalazione di eventi avversi standard, con etichette che specificano i requisiti di partecipazione del registro o il monitoraggio a lungo termine che tiene traccia degli effetti delle sperimentazioni cliniche non è riuscito a catturare completamente.
Sfide e soluzioni di etichettatura multilingue
Le aziende farmaceutiche che operano a livello globale devono affrontare sfide distinte nella creazione di etichette che soddisfino requisiti linguistici diversi, pur mantenendo la precisione e la conformità normativa.
Sfida: Qualità della traduzione e precisione culturale
La terminologia medica non si traduce direttamente nelle lingue, e il contesto culturale forma come i pazienti comprendono le informazioni sulla salute. Una traduzione tecnicamente accurata può completamente perdere come le persone in un mercato di destinazione effettivamente discutere concetti medici. I servizi di traduzione standard mancano delle esigenze di etichettatura farmaceutica specializzata della conoscenza.
Una traduzione efficace richiede specialisti che comprendano sia la terminologia medica che le aspettative normative di ogni mercato di destinazione. I traduttori hanno bisogno di fluenza nativa e di conoscenze di lavoro del settore farmaceutico per garantire precisione e contesto. La FDA e le altre agenzie di regolamentazione delineano quando i materiali tradotti necessitano di approvazione formale o di un traduttore certificato, con regole specifiche che variano da regione.
Sfida: Rispondere alle diverse esigenze linguistiche del mercato
Molti mercati impongono più lingue su un'unica etichetta. Il Canada richiede l'inglese bilingue e il francese. La Svizzera ha bisogno di tedesco, francese e italiano. Il Belgio richiede francese, olandese e tedesco. Adattare più traduzioni complete su etichette con vincoli spaziali rende difficili i compromessi tra la leggibilità e il rispetto dei requisiti normativi. Le aziende si rivolgono a questo lavoro lavorando con i designer di etichette specializzati in imballaggi farmaceutici multilingue, utilizzando tecniche come pieghe a fisarmonica, etichette opuscoli, o gerarchie di informazioni a livelli che danno la priorità alle informazioni di sicurezza critiche.
Sfida: Coordinare gli aggiornamenti tra lingue e mercati
Quando le informazioni sulla sicurezza cambiano sull'etichetta principale, ogni versione tradotta deve essere aggiornata. Raramente i tempi di regolamentazione dei mercati si allineano, creando periodi in cui i diversi paesi mostrano informazioni diverse sulla sicurezza sulle loro etichette.
La tecnologia fornisce soluzioni qui. I sistemi di memoria di traduzione mantengono la coerenza terminologica tra i prodotti, mentre i database terminologici assicurano che i termini tecnici si traducano uniformemente ogni volta che apparono. I sistemi di gestione dei flussi di lavoro riuniscono questo sistema tracciando quali versioni di etichette esistono in quali lingue, quali mercati hanno approvato ogni versione e dove sono necessari aggiornamenti. Questi sistemi aiutano le aziende a mantenere la conformità segnalando lacune e coordinando il controllo delle versioni nelle operazioni globali prima di problemi di superficie delle ispezioni normative.
Serializzazione e traccia & Requisiti traccia
I regolamenti di serializzazione hanno modificato il funzionamento dell'etichettatura farmaceutica richiedendo a ogni pacchetto di portare un identificatore unico che lo traccia attraverso la catena di approvvigionamento. Si tratta di affrontare problemi reali, tra cui la contraffazione e la necessità di eseguire rapidamente richiami quando emergono problemi di sicurezza. I regolamenti in tutto il mondo ora mandano la serializzazione, anche se i tempi di attuazione hanno variato per mercato. La legge statunitense sulla sicurezza della catena di fornitura dei farmaci richiedeva la conformità entro il 2023, mentre la direttiva sui medicinali falsificati dell'UE è entrata in vigore nel 2019. Altri mercati hanno seguito i propri programmi, creando un lancio globale sfalsato che richiedeva alle aziende di gestire contemporaneamente diverse esigenze. Questi regolamenti richiedono pacchetti per trasportare identificatori di prodotto con più data element - Codici nazionali di droga e numeri di serie unici insieme con i numeri di lotto e le date di scadenza - tutti in formati sia umani e macchine possono leggere.
La tecnologia dipende principalmente dai codici a barre 2D Data Matrix, che memorizzano grandi quantità di informazioni in un formato compatto. Questi codici compaiono in ogni fase della produzione, dalle linee di produzione e dai sistemi di controllo della qualità alle banche dati di distribuzione che tracciano ogni identificatore unico. Per la maggior parte dei team, la vera complessità è quando questi requisiti devono coesistere con etichette già imballate con contenuto obbligatorio. I codici a barre non possono oscurare il testo richiesto, e i numeri di serie devono apparire in entrambi i formati leggibili da una macchina e dall'altra. Allo stesso tempo, i dati di serializzazione devono rimanere nelle sue posizioni tradizionali per conformarsi ai processi di etichettatura farmaceutica di lunga data. In pratica, le aziende stanno progettando etichette che soddisfano due generazioni di aspettative normative, pur mantenendo chiarezza e leggibilità.
Controllo di qualità nell'etichettatura farmaceutica
Le revisioni del design delle etichette confrontano le etichette proposte con quelle approvate dalla regolamentazione. Il testo deve corrispondere esattamente, la grafica deve apparire come approvata, la formattazione deve seguire le specifiche. Anche le deviazioni minori richiedono documentazione e spesso fanno scattare la notifica normativa, motivo per cui queste revisioni stabiliscono la base per tutte le verifiche successive. Una volta che le prove del fornitore arrivano, gli strumenti di ispezione alimentati ad AI aiutano i team di garanzia della qualità a verificare che i file corrispondano ai documenti master approvati prima dell’inizio della stampa:
- Confronto Artwork verifica prove contro master approvati per catturare spostamenti di layout o elementi mancanti
- La verifica del codice a barre e del codice QR conferma che i dati codificati sono corretti e che ogni codice segue la formattazione richiesta prima della stampa
- Riconoscimento ottico dei caratteri (OCR) identifica le differenze di testo tra le prove e i file master approvati per garantire che non siano state introdotte modifiche indesiderate durante gli aggiornamenti
- Intended change detection Verifica se le correzioni annotate da versioni precedenti di file sono state applicate correttamente, i team di aiuto confermano che gli aggiornamenti richiesti sono stati implementati prima della produzione.
Questi sistemi funzionano continuamente durante le fasi di produzione, analizzare immediatamente le opere d'arte e il testo per segnalare i problemi piuttosto che scoprirli dopo che migliaia di unità sono state stampate. La sorveglianza umana gestisce le eccezioni e fornisce l'approvazione finale, ma l'ispezione automatizzata trasporta la maggior parte dei lavori di verifica su scala.
La documentazione lega tutto insieme, dimostrando alle autorità di regolamentazione che le procedure sono state effettivamente seguite. I record di lotto catturano ciò che accade durante la produzione - quali ispezioni hanno funzionato, quali risultati sono tornati, come le deviazioni sono state risolte, chi ha approvato il rilascio. Durante le ispezioni normative, questi registri dimostrano che il vostro sistema di controllo della qualità funziona effettivamente piuttosto che solo esistente come procedure scritte. La documentazione mancante crea problemi di conformità anche quando le etichette stesse sono corrette, perché i regolatori non possono verificare il processo lavorato come progettato. Aziende che integrano queste misure di controllo della qualità dalla progettazione alla documentazione cattura errori di etichettatura prima che diventino problemi di mercato, mentre coloro che si affidano a controlli reattivi scoprono problemi durante gli audit quando le correzioni sono molto più costose.
Strumenti di automazione per l'etichettatura conforme
Queste piattaforme gestiscono flussi di lavoro in cui più team esaminano le etichette prima della produzione, garantendo un'etichettatura conforme in ogni fase della revisione. I contenuti passano dalla ricerca allo sviluppo dei prodotti alla revisione normativa, poi alle fasi di progettazione e marketing per il layout, verifica del marchio e delle richieste prima di raggiungere la garanzia della qualità per i controlli finali di accuratezza. Il sistema segue che cosa e quando, creando la pista di controllo della domanda di ispezioni. Le aziende possono scalare questi flussi di lavoro oltre che le catene di posta elettronica e unità condivise gestiscono.
La convalida segue lo stesso rigore richiesto per qualsiasi sistema di fabbricazione farmaceutica. Il processo inizia con la qualificazione di installazione per confermare la corretta installazione, passa attraverso la qualificazione operativa che verifica tutte le funzioni, e si conclude con la qualificazione delle prestazioni che dimostra la coerenza dell’accuratezza durante le normali operazioni. La convalida dimostra che il sistema automatizzato impedisce in modo affidabile gli errori di etichettatura piuttosto che introdurne di nuovi.
L'integrazione con i sistemi di gestione delle informazioni normative collega i contenuti approvati direttamente alla produzione di etichette. Gli aggiornamenti alle informazioni di prescrizione dopo le approvazioni della FDA fluiscono direttamente alle etichette pertinenti senza che nessuno copii manualmente il testo tra i sistemi, che taglia gli errori di trascrizione mentre accelera i cicli di cambiamento. I principali vantaggi includono aggiornamenti più rapidi delle etichette e un controllo più rigoroso delle versioni in tutti i siti globali, supportati da flussi di lavoro strutturati di revisione che rafforzano la conformità normativa generale. Le aziende vedono i guadagni di efficienza operativa riducendo i touchpoint manuali e ottenendo visibilità in tempo reale nello stato di etichettatura attraverso le operazioni di produzione.
Migliori prassi per le comunicazioni regolamentari
Coinvolgere le questioni di regolamentazione durante lo sviluppo del prodotto in modo che considerazioni di etichettatura possono modellare come progettare le sperimentazioni cliniche e raccogliere dati. Attendere fino al tempo di archiviazione significa che sei bloccato cercando di rendere i vostri dati di supporto etichettatura sostiene che non è stato progettato per dimostrare.
La tua sottomissione deve essere etichettata nel formato che l'agenzia si aspetta. La FDA vuole il formato labeling (SPL) strutturato, uno standard basato su XML i loro sistemi possono elaborare elettronicamente. Contenuto segue gli standard del settore - sezioni appaiono in ordine prescritto con tutti gli elementi richiesti inclusi. Ottenere le specifiche tecniche sbagliate e la vostra sottomissione viene rifiutata sopra la formattazione piuttosto che qualsiasi reale sicurezza o problemi di efficacia.
Tracciando ciò che è cambiato dalla tua versione attualmente approvata separa gli aggiornamenti di etichettatura fluidi da mal di testa normativi. Alcuni cambiamenti necessitano di approvazione preventiva prima dell'attuazione, altri passano sotto le disposizioni "modifiche in corso" e alcuni vengono appena riportati annualmente. Mescolare questi e stai creando problemi di conformità che rallentano ottenere aggiornamenti sul mercato. Le comunicazioni elettroniche hanno sostituito la carta, con il Gateway della FDA elaborando contenuti strutturati in modo diverso rispetto ai PDF. Molte aziende portano in servizi specializzati per gestire i requisiti tecnici e garantire l'accuratezza. Dopo l'approvazione, i vostri sistemi devono garantire che solo le versioni approvate raggiungano la produzione. Cambia il controllo che effettivamente funziona impedisce alle vecchie etichette di scivolare attraverso dopo che le nuove versioni vengono approvate.
Requisiti di formazione e documentazione
Tutti coloro che sono coinvolti nelle operazioni di etichettatura devono capire cosa stanno facendo e perché importa, e devi provarlo. Queste sessioni non sono eventi una tantum. La formazione continua mantiene i team attuali con cambiamenti normativi e nuovi sistemi. La vera misura non è se l'addestramento accade, ma se le squadre capiscono i requisiti attuali quando gli ispettori arrivano.
Le procedure operative standard (SOP) documentano come la tua azienda gestisce ogni fase del processo di etichettatura, mostrando regolatori si segue un processo definito piuttosto che prendere decisioni in modo diverso a seconda di chi sta lavorando quel giorno. I POS devono coprire:
- Come i file artwork vengono gestiti e controllati dalla versione
- Come le approvazioni fluiscono attraverso l'organizzazione
- Come le modifiche vengono iniziate, riviste e implementate
- Come deviazioni e problemi vengono indagati e risolti
Scrivere SOPs è semplice; seguirli costantemente e aggiornarli quando le pratiche di etichettatura cambiano è dove le aziende lottano. I record dimostrano che il vostro sistema di qualità funziona come progettato, non solo su carta. Essi devono dimostrare che il personale formato ha seguito le procedure approvate, che le ispezioni sono state completate come previsto, e che eventuali deviazioni sono state adeguatamente indagate. Durante le ispezioni normative, questi fascicoli rispondono alla domanda più fondamentale: avete effettivamente fatto quello che le vostre procedure dicono di fare? La formazione va oltre gli specialisti di etichettatura a chiunque tocca il processo, dal personale di regolamentazione che scrive i contenuti ai grafici che creano opere d'arte al personale di garanzia di qualità che approva le corse di produzione. I team che sottolineano i programmi di formazione forti tendono ad avere meno problemi di conformità durante gli audit e possono rispondere più rapidamente quando i requisiti cambiano. L'alternativa sta scoprendo le lacune quando le autorità di regolamentazione li trovano, che costa molto di più da risolvere.
Tendenze future dell'etichettatura farmaceutica
L'etichettatura digitale sta andando oltre le versioni PDF degli inserti di carta. I codici QR sui pacchetti ora si collegano direttamente alle informazioni di prescrizione che aggiornano in tempo reale, consentire agli operatori sanitari di accedere ai dati di sicurezza attuali piuttosto che fare affidamento su inserti stampati che potrebbero essere vecchi di mesi. La FDA riconosce che le etichette cartacee hanno limiti, in particolare per i prodotti complessi in cui le informazioni complete superano ragionevoli vincoli di spazio, ma i regolatori non hanno abbandonato l'etichettatura fisica. Le informazioni fondamentali sulla sicurezza richiedono ancora la presenza sull'imballaggio per garantire l'accesso senza dispositivi digitali.
La pressione ambientale sta rimodellando le decisioni sugli imballaggi in modo talvolta in contrasto con i requisiti normativi. Il settore farmaceutico deve far fronte a esigenze di riduzione dei rifiuti e di utilizzo di materiali sostenibili, obiettivi che possono scontrarsi con normative che specificano particolari costruzioni di imballaggio. L'Unione europea ha imposto requisiti di imballaggio particolarmente aggressivi e sostenibili che incidono sulla progettazione e sulla produzione delle etichette.
Le etichette intelligenti con chip NFC o tecnologie embedded verificano l'autenticità del prodotto durante il monitoraggio dell'uso e il monitoraggio dell'esposizione alla temperatura durante tutta la distribuzione. Queste capacità sollevano nuove questioni normative sulla privacy e la sicurezza dei dati, nonché su come la funzionalità delle etichette intelligenti si estenda al territorio dei dispositivi medici. La tecnologia esiste; il quadro normativo in materia è ancora in forma.
Risorse per i professionisti dell'etichettatura farmaceutica
Le associazioni professionali come la Regulatory Affairs Professionals Society (RAPS) e la Drug Information Association (DIA) ti collegano a persone che affrontano simili sfide di etichettatura attraverso conferenze e gruppi di lavoro. Si ottiene una panoramica su come i regolatori affrontano i nuovi requisiti mentre si vede come altre aziende risolvono i problemi che si stanno affrontando. RAPS offre anche la certificazione degli affari normativi che copre le competenze di etichettatura, mentre i programmi di formazione specializzati affrontano le lacune specifiche quando i team hanno bisogno di ottenere corrente su nuove normative o tecnologie.
Il supporto esterno proviene da due direzioni. Consulenza aziende portare competenze normative per la navigazione di sottomissioni complesse, in particolare quando si tratta di requisiti sconosciuti o scadenze strette. I fornitori di tecnologia offrono piattaforme che gestiscono la gestione delle versioni nei mercati coordinando le modifiche e le approvazioni. La scelta tra di loro dipende spesso dal fatto che la vostra sfida è capire cosa vogliono i regolatori o gestire la complessità operativa di farla fare.
Per le squadre che gestiscono contenuti high-stakes, la precisione non è opzionale. GlobalVison’s Verify dà ai team la certezza che ogni versione approvata è pronta per l'ispezione.
