
Un vial mal etiquetado se desliza por cada puesto de control y llega a una farmacia hospitalaria. Las instrucciones de la dosis parecen rutinas: un solo dígito que nadie nota hasta que es demasiado tarde. Es un claro recordatorio de que el etiquetado farmacéutico compatible sigue siendo una de las salvaguardas más críticas de la industria farmacéutica.
Introducción al Cumplimiento de Etiquetado Físico
El etiquetado farmacéutico conforme sirve de línea directa entre las compañías farmacéuticas y los profesionales de la salud, proporcionando información esencial que afecta directamente a la seguridad del paciente. Esto incluye todo lo que se ve en los envases y contenedores de medicamentos junto con los materiales que lo acompañan. asegurando que los profesionales médicos y los pacientes tengan información precisa para tomar decisiones de tratamiento seguras e informadas.
Los organismos reguladores de todo el mundo mantienen directrices estrictas porque los errores de etiquetado pueden causar daños graves. En Estados Unidos, la Food and Drug Administration (FDA) define y aplica los requisitos de etiquetado farmacéutico tanto para medicamentos de prescripción como para dispositivos médicos. En toda la Unión Europea, este papel corresponde a la Agencia Europea de Medicamentos (EMA). En coordinación con Health Canada, la Agencia Japonesa de Fiduticos y Dispositivos Médicos (PDMA), y Administración de Productos Therapeutic (TGA), estas agencias reguladoras crean marcos diseñados para proteger la salud pública y mantener la consistencia en los mercados. Los esfuerzos para armonizar las normas a través del Consejo Internacional para la Armonización (ICH) continuan, pero la alineación total sigue fuera de alcance y cada región aplica sus propios requisitos reglamentarios que las empresas farmacéuticas deben gestionar con cuidado. Mientras tanto, el panorama regulatorio sigue evolucionando: la entrega de información digital está cambiando la forma en que los proveedores de atención sanitaria acceden a los datos, y las maniobras de serialización están redefiniendo cómo funcionan los procesos de etiquetado farmacéutico en toda la industria.
Llave de estructuras regulatorias para el etiquetado utical
Since 2006, the FDA’s Physician Labeling Rule (PLR) has defined the modern standard for prescription drug labeling, bringing consistency and structure to every approved product. Introdujo una clara jerarquía para prescribir información, con una sección concisa de "Resaltados" para profesionales de la salud y normas específicas sobre cómo debe aparecer el contenido restante. Estas regulaciones responden directamente a los errores de medicamentos que ocurren cuando la información crítica queda enterrada en un texto denso. Estos reglamentos estrictos se aplican en todas las categorías de productos, incluyendo los medicamentos contra los que los consumidores tienen acceso sin recetas.
Los estándares internacionales añaden niveles de requisitos para las empresas que operan a través de los mercados. Los requisitos Resumen de Características de Productos (SmPC) y Folleto de Paquetes difieren de los formatos FDA tanto en la estructura como en el énfasis del contenido. Si bien las directrices del ICH tienen como objetivo armonizar los requisitos técnicos, cada región mantiene enfoques distintos. La siguiente tabla muestra cómo los principales mercados manejan los requisitos de etiquetado clave de manera diferente.
Requisitos regulatorios de etiquetado en los principales mercados
| Requisito | FDA (U.S.) | EMA (EU) | Canadá de salud |
|---|---|---|---|
| Formato de información de receta | Formato PLR con sección de Resaltados seguido de Información de Recarga Completa | PPC con secciones estandarizadas | Monográfico de producto con tres partes: Parte I (profesional de la salud), Parte II (paciente), Parte III (científico) |
| Secciones/orden requeridos | Formato estructurado con secciones requeridas por pautas de PLR | Estandarizado plantilla en el mapaPC con secciones obligatorias en el orden especificado | Formato estructurado siguiendo la guía de Health Canada con los elementos requeridos en cada parte |
| Actualizar requerimientos de notificación | Anterior aprobación de cambios de eficacia; CBE para actualizaciones de seguridad; informes anuales para cambios menores | Procedimientos de variación con diferentes rutas de aprobación y cronogramas basados en el significado del cambio | Sistema de notificación basado en la importancia del cambio y el nivel de riesgo |
| Normas de formato electrónico | SPL (Etiqueta de producto estructurada) en formato XML requerido para todos los envíos | formato eCTD con módulos específicos para etiquetar contenido | Formato eCTD requerido; envíos bilingües en plantillas estandarizadas |

Japón mandates que todo el contenido de la etiqueta se escriba en japonés y formatea para que coincida con las prácticas de documentación médica local. El Programa de Ventajas de Ventajas de Ventajas de Ventajas de Ventajas de Ventajas de Ventajas de Ventajas de Ventajas de Ventajas de Ventajas de Ventajas de Ventajas de Ventajas de Ventajas de Ventajas de Ventajas de Ventajas de Ventajas de Ventajas de Ventajas de Ventajas de Ventajas de Ventajas de Ventaja. Las empresas que lanzan productos farmacéuticos en todo el mundo deben mantener múltiples versiones de etiquetas, cada una de las cuales cumple con las normas reguladoras locales al mismo tiempo que garantiza la consistencia en mensajes de seguridad básicos. Gestionar control de versiones en estos mercados requiere sistemas que rastreen qué etiqueta se aplica la versión dónde, hacer que pharma etiquete el cumplimiento de una disciplina que combine experiencia en asuntos regulatorios con precisiones operacionales para navegar por el laberinto regulatorio global de hoy.
Componentes esenciales de las etiquetas de fármacos conformes
Las etiquetas de fármacos de prescripción proporcionan información diferente dependiendo del lugar en el que aparezcan. La etiqueta inmediata del contenedor - lo que está en la botella o en el vial - cubre lo básico: nombre de la droga, dosificación, fuerza, ruta de administración, y número de lote. La etiqueta Carton proporciona más espacio con el que trabajar. El panel principal de visualización muestra el nombre y la fuerza establecidos junto con la cantidad de paquetes. Los paneles laterales manejan los puntos destacados de la información de la prescripción y los detalles de la orientación del paciente, además de la información del fabricante. Las reglamentaciones especifican dónde aparece todo y cómo de forma clara, hasta tamaños de fuentes exactos que hacen de la etiqueta de producto una disciplina que exacerba.
Comienza con destacar que la información superficial crítica de la prescripción y la seguridad, luego se mueve a través de la información completa de la prescripción. Cada sección sigue un formato prescrito y cualquier desviación requiere justificación y aprobación reglamentaria. La estructura mantiene los datos de seguridad esenciales consistentes y fáciles de encontrar, ayudando a los revisores y profesionales de la salud a acceder rápidamente a los detalles correctos. Las secciones clave incluyen:
- Indicaciones y uso
- Dosaje y administración
- Contraindicaciones
- Advertencias y precauciones
- Reacciones adversas
- Interacciones con drogas
- Utilizar en poblaciones específicas
- Farmacología clínica
Las secciones de información de pacientes deben alcanzar un nivel de lectura de sexto a octavo grado en los Estados Unidos, tal como se describe en las directrices de legibilidad de la FDA. Otras agencias reguladoras pueden aplicar diferentes estándares de alfabetización o formato, reflejando el contexto lingüístico y sanitario en sus regiones. Las guías de medicación para medicamentos con riesgos graves ayudan a los pacientes a entender el uso seguro sin abrumarlos con detalles técnicos. Las instrucciones claras de uso e instrucciones de dosis indican a los pacientes cómo tomar sus medicamentos correctamente y qué cuidar.
Entendiendo el Panel de Información sobre Drogas
El panel de información sobre medicamentos proporciona a los profesionales de la salud los detalles que necesitan para recetar y administrar los medicamentos de forma segura. Dentro de ese contenido, las instrucciones de almacenamiento tienen un peso especial porque determinan si un medicamento mantiene su estabilidad y eficacia a lo largo de su vida útil. Para proteger esa estabilidad, las etiquetas definen los rangos de temperatura requeridos y explican cómo proteger los productos de la luz y la humedad durante el almacenamiento o el transporte. Cuando esos límites se pasan por alto, la degradación puede ocurrir antes de que un producto llegue a los pacientes, una pérdida de calidad evitable que subraya la necesidad de una revisión exhaustiva de la calidad de los productos. Algunos medicamentos deben ser ignífugos, otros protegidos de la luz y muchos pierden la niebla si están expuestos a la humedad en la transita. Estos requisitos determinan si los productos siguen siendo efectivos o si se convierten en sustancias inútiles (o nocivas). Cuando estas instrucciones se comunican de forma poco clara o se ignoran por completo, los productos pierden eficacia o se vuelven potencialmente dañinos.
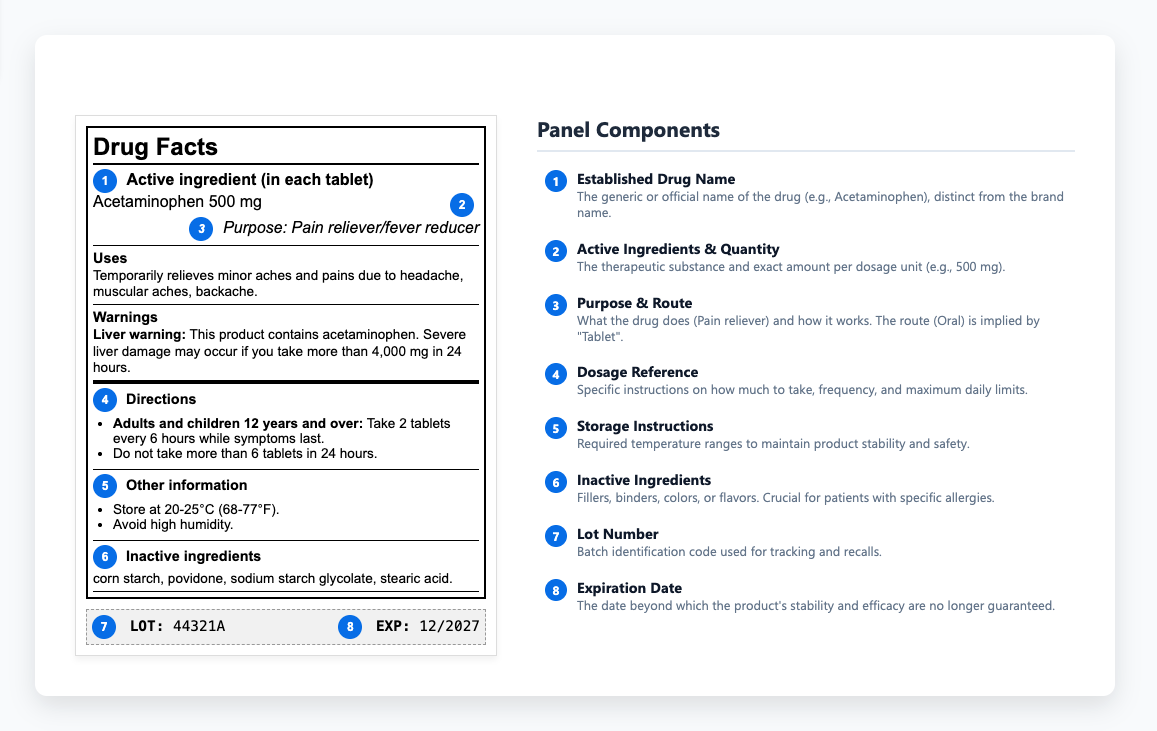
Cuando las formulaciones cambian o aparecen nuevos datos de seguridad, las actualizaciones de la etiqueta a veces se quedan rezagadas, creando discordancias entre envíos aprobados y las etiquetas liberadas al mercado. Esas incoherencias pueden parecer menores al principio, pero a menudo surgen durante las inspecciones regulatorias, ralentizando las revisiones de cumplimiento y añadiendo riesgos innecesarios. Cuentas comparan el contenido de la etiqueta directamente con las versiones aprobadas, por lo que estos problemas rara vez permanecen ocultos durante mucho tiempo.
Información de seguridad y etiquetas de advertencia
Las advertencias sobre cajas negras son la forma más grave de alerta de seguridad de la FDA, usado para enfatizar los riesgos graves o peligrosos que deben aparecer de forma destacada en las etiquetas de medicamentos de prescripción médica. Se muestran cuando las precauciones estándar no previenen daños graves. La FDA los añade a partir de datos posteriores al mercado que revelan patrones de reacciones adversas peligrosas, a veces incluidas las muertes. Estas advertencias se sitúan en la cima de la información recetada en audaces fronteras, utilizando un lenguaje directo sobre cuál es el riesgo y quién es más vulnerable. Las Contraindicaciones van un paso más allá al identificar cuándo la droga no debe ser utilizada en absoluto. Esto incluye interacciones peligrosas con otros medicamentos, riesgos documentados de embarazo y condiciones médicas preexistentes que harían que el medicamento resultara inseguro.
La información de reacción publicitaria abarca efectos leves y esperados hasta complicaciones raras pero graves. Presentar datos de frecuencia junto con esos resultados ayuda a situar el nivel de riesgo en contexto para pacientes y revisores. Para reducir ese riesgo, las estrategias de mitigación a menudo incluyen ajustar las dosis para poblaciones específicas, aplicar protocolos de monitoreo y guiar a los proveedores de servicios de salud en el asesoramiento y el seguimiento apropiados. Para algunos medicamentos de alto riesgo, los programas Evaluación de Riesgo y Mitigación (REMS) exigen elementos de etiquetado adicionales para garantizar la seguridad del paciente como requisitos vinculantes.

Requisitos de Etiquetado Especializado para medicamentos genéricos
El etiquetado de medicamentos genéricos tiene que coincidir con la etiqueta del producto de marca casi exactamente, un requisito que la FDA obliga a garantizar que los genéricos ofrezcan la misma seguridad y eficacia que el original. Puedes diferir en algunas cosas: la información de patentes queda fuera ya que pertenece a la marca, mientras que los detalles de su propio fabricante y los códigos NDC reemplazan los suyos. La sección "Cómo Suministrar" también puede reflejar su embalaje específico.
El trabajo real comienza cuando cambia la etiqueta de marca. Una nueva advertencia o contraindication se añade en base a los datos posventa y su etiqueta genérica debe coincidir. La ruta de "Cambios Efectos" de la FDA te permite hacer ciertas actualizaciones de seguridad sin esperar la aprobación, pero sigue teniendo la responsabilidad de captar estos cambios y actuar en consecuencia. Las empresas que confían en que los reguladores les informen cuándo las etiquetas están obsoletas terminan atrasadas, La solución de problemas después de las inspecciones reglamentarias los marcan en lugar de seguir adelante. Mantener corriente significa rastrear cuando los productos de referencia cambian, luego evaluar cómo estas actualizaciones afectan a sus productos específicos. Las actualizaciones deben desplegarse a través de los sitios de fabricación con documentación que demuestre el cumplimiento durante las auditorías. La presión aumenta cuando las actualizaciones de seguridad requieren medidas inmediatas, pero sus procesos normales tardan meses. Los fabricantes de genéricos que manejan este pozo construyen sistemas de monitoreo que capturan los cambios temprano en lugar de esperar a que surjan problemas durante las revisiones de cumplimiento.
Productos biológicos: Consideraciones Específicas de Etiquetado
Los productos biológicos provienen de los órganos vivos en lugar de de la síntesis química, que cambia lo que debe aparecer en las etiquetas. La biología requiere detalles sobre su fuente biológica y sus procesos de fabricación, junto con advertencias sobre las respuestas del sistema inmunitario que los medicamentos químicos no activan.
Diferencias clave de etiquetado
| Elemento de Etiquetado | Drogas pequeñas moléculas | Productos biológicos |
|---|---|---|
| Detalles de manipulación | Descripción general del proceso | Fuente biológica detallada, información de la línea celular y controles de proceso que afectan a las características del producto |
| Requisitos de almacenamiento | A menudo la temperatura ambiente estable con especificaciones estándar | Típicamente requieren una ligereza con rangos estrechos de temperatura; muchos necesitan protección de la luz y son sensibles a la agitación |
| Información de inmunogenicidad | No aplicable | Alertas requeridas sobre posibles respuestas inmunitarias y desarrollo de anticuerpos antidrogas |
| Relación biosimilar | No aplicable | Debe incluir una declaración que explique la relación a referencia biológica y naturaleza de la similitud |
| Vigilancia posmercado | Reporte estándar de eventos adversos | Programas de monitorización mejorados con participación específica en el registro o requerimientos de seguimiento a largo plazo |
Los requisitos de almacenamiento para la biología van más allá de las especificaciones estándar, ya que estos productos se degradan fácilmente en condiciones que no afectarían a los medicamentos químicos. La mayoría de las personas necesitan refrigeración dentro de rangos de temperatura ajustados, y una exposición breve al calor, a la luz o a la agitación puede hacerlos ineficaces o potencialmente dañinos. Las etiquetas describen cómo deben almacenarse y transportarse los productos, detallando los límites de temperatura y los pasos de manejo confirmados durante el desarrollo.
Las advertencias sobre inmunogenicidad explican cómo el sistema inmunitario puede reaccionar a los productos biológicos. Las etiquetas explican el riesgo de desarrollar anticuerpos antifármacos y resumen la manera en que los ensayos clínicos evaluaron ese riesgo, incluyendo lo que los resultados significan para los pacientes. Los proveedores de atención médica utilizan esta información para monitorear la respuesta al tratamiento y ajustar la terapia cuando se desarrollan reacciones inmunitarias. La vigilancia posterior al mercado se extiende más allá de la notificación estándar de eventos adversos, con etiquetas que especifican los requisitos de participación en el registro o la supervisión a largo plazo que rastrea los efectos de los ensayos clínicos no pudieron capturarse completamente.
Desafíos y soluciones de etiquetado multilingües
Las empresas farmacéuticas que operan en todo el mundo se enfrentan a desafíos distintos a la hora de crear etiquetas que cumplan con los diversos requisitos lingüísticos y mantengan la precisión y la conformidad regulatoria.
Desafío: calidad de la traducción y precisión cultural
La terminología médica no se traduce directamente a través de los idiomas, y el contexto cultural da forma a cómo los pacientes entienden la información sobre la salud. Una traducción técnicamente precisa puede perder completamente la manera en que la gente en un mercado objetivo habla realmente de conceptos médicos. Los servicios de traducción estándar carecen de las exigencias de etiquetado farmacéutico especializado en conocimientos.
La traducción efectiva requiere de especialistas que entiendan tanto la terminología médica como las expectativas reguladoras de cada mercado objetivo. Los traductores necesitan fluidez y conocimientos nativos del dominio farmacéutico para garantizar la precisión y el contexto. La FDA y otras agencias reguladoras describen cuando los materiales traducidos necesitan una aprobación formal o un traductor certificado, con reglas específicas que varían según la región.
Desafío: Cumplir con diversos requisitos de idioma del mercado
Muchos mercados mandan múltiples idiomas en una sola etiqueta. Canadá requiere el inglés bilingüe y el francés. Suiza necesita alemán, francés e italiano. Bélgica necesita francés, Dutch y alemán. La introducción de múltiples traducciones completas en etiquetas con limitaciones espaciales obliga a difíciles compensaciones entre legibilidad y cumplimiento de los requisitos reglamentarios. Las empresas abordan esta cuestión trabajando con diseñadores de etiquetas especializados en envases farmacéuticos multilingües, el uso de técnicas como pliegues de accordión, etiquetas de libros o jerarquías de información escalada que prioricen la información de seguridad crítica.
Desafío: Coordenando actualizaciones entre idiomas y mercados
Cuando la información de seguridad cambia en la etiqueta maestra, cada versión traducida necesita actualizarse. Los plazos regulatorios en los mercados rara vez alientan, creando períodos en los que diferentes países muestran información de seguridad diferente en sus etiquetas.
La tecnología ofrece soluciones a este respecto. Los sistemas de memoria de traducción mantienen la coherencia terminológica entre los productos, mientras que las bases de datos de terminología garantizan que los términos técnicos se traduzcan de forma uniforme cada vez que aparecen. Los sistemas de gestión de flujo de trabajo unen esto rastreando qué versiones de etiqueta existen en los idiomas, que mercados han aprobado cada versión y donde se necesitan actualizaciones. Estos sistemas ayudan a las empresas a mantener el cumplimiento al marcar brechas y coordinar el control de versiones a través de operaciones globales antes de que las inspecciones regulatorias presenten problemas superficiales.
Serialización y seguimiento de los requerimientos & seguimiento
Las regulaciones de serialización cambiaron la forma en que funciona el etiquetado farmacéutico al exigir que cada paquete llevara un identificador único que lo rastreara a través de la cadena de suministro. Esto aborda problemas reales, como la falsificación y la necesidad de ejecutar retiradas rápidas cuando surjan problemas de seguridad. Regulaciones en todo el mundo ahora mandan serialización, aunque los plazos de implementación han variado según el mercado. La Ley de Seguridad de la Cadena de Abastecimiento de Drogas de los Estados Unidos exigió el cumplimiento para 2023, mientras que la Directiva sobre medicamentos falsificados de la UE entró en vigor en 2019. Otros mercados siguieron sus propios programas, creando un despliegue global escalonado que obligó a las empresas a gestionar los diferentes requisitos simultáneamente. Estas regulaciones requieren paquetes para llevar los identificadores de producto con múltiples elementos de datos: Códigos nacionales de medicamentos y números de serie únicos junto con números de lote y fechas de caducidad- todo en formatos tanto humanos como máquinas pueden leer.
La tecnología depende principalmente de códigos de barras 2D Data Murgx, que almacenan grandes cantidades de información en un formato compacto. Estos códigos aparecen en todas las fases de producción, desde líneas de fabricación y sistemas de control de calidad hasta bases de datos de distribución que rastrean cada identificador único. Para la mayoría de los equipos, la complejidad real viene cuando estos requisitos deben coexistir con etiquetas ya llenas de contenido obligatorio. Los códigos de barras no pueden ocultar el texto requerido, y los números de serie deben aparecer tanto en formatos legibles por el ser humano como por la máquina. Al mismo tiempo, los datos de serialización deben permanecer en sus posiciones tradicionales para cumplir con los antiguos procesos de etiquetado farmacéutico. En la práctica, las empresas están diseñando etiquetas que satisfacen dos generaciones de expectativas regulatorias y mantienen la claridad y la legibilidad.
Control de calidad en el etiquetado utical
Las revisiones de diseño de etiquetas comparan las etiquetas propuestas con las presentaciones reglamentarias aprobadas. El texto tiene que coincidir exactamente, los gráficos deben aparecer como aprobados, el formato tiene que seguir las especificaciones. Incluso las desviaciones menores requieren documentación y a menudo desencadenan notificaciones regulatorias, razón por la cual estas revisiones establecen la línea de referencia para todas las verificaciones posteriores. Una vez que lleguen las pruebas del proveedor, las herramientas de inspección alimentadas por AIs ayudan a los equipos de aseguramiento de calidad entrantes a verificar que los archivos coinciden con los documentos maestros aprobados antes de que comience la impresión:
- comparación de Artwork comprueba las pruebas contra maestros aprobados para capturar turnos de diseño o elementos faltantes
- Barcode and QR code verification confirms that encoded data is correct and that each code follows required formatting before printing
- Optical character recognition (OCR) identifies text differences between proofs and approved master files to ensure no unintended changes were introduced during updates
- Intended change detection checks whether annotated corrections from earlier file versions were applied correctly, helping teams confirm that requested updates were implemented before production.
Estos sistemas funcionan continuamente durante las operaciones de producción, analizando el diseño y el texto para marcar los problemas inmediatamente en lugar de descubrirlos después de que se hayan imprimido miles de unidades. El ser humano maneja excepciones y proporciona la aprobación final, pero la inspección automatizada conlleva la mayor parte del trabajo de verificación a escala.
La documentación vincula todo entre sí, demostrando a los reguladores que los procedimientos se han seguido realmente. Los registros de Batch capturan lo que sucede durante la producción - qué inspecciones ejecutaron, qué resultados volvieron, cómo se resolvieron las desviaciones, quién aprobó la liberación. Durante las inspecciones regulatorias, estos registros demuestran que su sistema de control de calidad funciona realmente más que como procedimientos escritos. La documentación que falta crea problemas de cumplimiento incluso cuando las etiquetas son correctas, porque los reguladores no pueden verificar el proceso que funcionó como está diseñado. Las empresas que integran estas medidas de control de calidad desde el diseño hasta la documentación capturan errores de etiquetado antes de que se conviertan en problemas de mercado. mientras que aquellos que dependen de las comprobaciones reactivas descubren problemas durante las auditorías cuando las correcciones son mucho más caras.
Herramientas de automatización para Etiquetado Conformante
Estas plataformas gestionan flujos de trabajo en los que varios equipos revisan las etiquetas antes de la producción, asegurando un etiquetado compatible en cada etapa de revisión. El contenido pasa de la investigación y el desarrollo de productos a la revisión regulatoria, luego a las fases de diseño y marketing para el diseño, verificación de marcas y reclamaciones antes de llegar a la garantía de calidad para comprobaciones de exactitud finales. El sistema hace un seguimiento de quién revisó qué y cuándo, creando la demanda de inspecciones regulatorias del control de auditoría. Las empresas pueden escalar estos flujos de trabajo más allá de lo que manejan las cadenas de correo electrónico y las unidades compartidas.
La validación sigue el mismo rigor necesario para cualquier sistema de fabricación farmacéutica. El proceso comienza con la calificación de instalación para confirmar la correcta configuración, se mueve a través de la calificación operativa que prueba todas las funciones, y concluye con una calificación de rendimiento que demuestre una precisión consistente durante las operaciones normales. La validación demuestra que el sistema automatizado evita de forma fiable los errores de etiquetado en lugar de introducir los nuevos.
La integración con los sistemas de gestión de información regulatoria conecta el contenido aprobado directamente a la producción de etiquetas. Las actualizaciones de la información de prescripción después de que las aprobaciones de la FDA fluyen directamente a etiquetas relevantes sin que nadie copie manualmente texto entre sistemas, que corta los errores de transcripción al acelerar los ciclos de cambio. Los principales beneficios incluyen actualizaciones de etiquetas más rápidas y control de versiones más riguroso a través de sitios globales, soportados por flujos de trabajo de revisión estructurados que fortalecen el cumplimiento general de la normativa. Las empresas ven mejoras en la eficiencia operacional reduciendo los puntos de contacto manuales y obteniendo visibilidad en tiempo real en el estado de etiquetado a través de las operaciones de fabricación.
Mejores prácticas para los envíos regulatorios
Involucre los asuntos regulatorios durante el desarrollo del producto de modo que las consideraciones de etiquetado puedan dar forma a cómo diseñar ensayos clínicos y recopilar datos. Esperar hasta que se registre el tiempo significa que estás atascado al intentar hacer que tus datos soporten las declaraciones de etiquetas no estén diseñadas para probar.
Su presentación necesita etiquetar en el formato que la agencia espera. La FDA quiere un formato estructurado etiquetado de producto (SPL), un estándar basado en XML que sus sistemas pueden procesar electrónicamente. El contenido sigue los estándares de la industria - las secciones aparecen en orden prescrito con todos los elementos requeridos incluidos. Infórmate de las especificaciones técnicas y tu presentación es rechazada por el formato en lugar de cualquier preocupación real de seguridad o eficacia.
El seguimiento de lo que cambió de su versión aprobada actualmente separa las actualizaciones de etiquetas suaves de los dolores de cabeza regulatorios. Algunos cambios necesitan aprobación previa antes de la implementación, otros pasan por debajo de las disposiciones de "cambios que se están realizando", y algunos simplemente se informan anualmente. Mezcla estos y estás creando problemas de cumplimiento que ralentizan las actualizaciones en el mercado. Los envíos electrónicos han reemplazado el papel, por el contenido estructurado de la pasarela de la FDA que procesa el contenido de forma diferente a los PDFs. Muchas empresas ofrecen servicios especializados para gestionar los requisitos técnicos y garantizar la exactitud. Después de la aprobación, sus sistemas deben garantizar que sólo las versiones aprobadas llegan a la producción. Cambie el control que realmente funciona evita que las etiquetas antiguas se deslizan después de que se aprueben las nuevas versiones.
Requisitos de formación y documentación
Todos los involucrados en las operaciones de etiquetado deben entender lo que están haciendo y por qué importa, y es necesario que lo prueben. Estas sesiones no son eventos únicamente. La formación continua mantiene a los equipos actualizados con los cambios regulatorios y los nuevos sistemas. La verdadera medida no es si la formación se produce, sino si los equipos entienden los requisitos actuales cuando llegan los inspectores.
Procedimientos operativos estándar (SOPs) documentan cómo su empresa maneja cada paso del proceso de etiquetado, mostrando a los reguladores que sigues un proceso definido en lugar de tomar decisiones de forma diferente dependiendo de quién esté trabajando ese día. Los SO necesitan cubrir:
- Cómo se gestionan los archivos de artwork y se controla la versión
- Cómo fluyen las aprobaciones a través de la organización
- Cómo se inician, revisan e implementan los cambios
- Cómo se investigan y resuelven las desviaciones y problemas
Escribir SOP es sencillo; seguirlos consistentemente y actualizarlos cuando cambian las prácticas de etiquetado es donde las empresas luchan. Los registros demuestran que su sistema de calidad funciona como está diseñado, no sólo sobre el papel. Deben demostrar que el personal capacitado siguió los procedimientos aprobados, las inspecciones se completaron según lo previsto, y cualquier desviación se investigó adecuadamente. Durante las inspecciones regulatorias, estos archivos responden a la pregunta más básica: ¿realmente hizo lo que sus procedimientos le dicen hacer? La capacitación se extiende más allá de los especialistas etiquetados a cualquiera que toque el proceso, Desde el personal regulador que escribe contenido a diseñadores gráficos que crean obras de arte hasta personal de aseguramiento de la calidad que apruebe corridas de producción. Los equipos que enfatizan los fuertes programas de capacitación tienden a tener menos problemas de cumplimiento durante las auditorías y pueden responder más rápidamente cuando cambian los requisitos. La alternativa es descubrir lagunas cuando los reguladores las encuentran, lo que cuesta mucho más arreglarlo.
Tendencias futuras en etiquetado farmacéutico
El etiquetado digital va más allá de las versiones PDF de las inserciones de papel. Los códigos QR en los paquetes ahora enlazan directamente a la información que se actualiza en tiempo real permitir que los profesionales de la salud accedan a los datos de seguridad actuales en lugar de depender de los insertos impresos que podrían tener meses de edad. La FDA reconoce que las etiquetas de papel tienen límites, particularmente para productos complejos donde la información completa excede las restricciones razonables de espacio, pero los reguladores no han abandonado el etiquetado físico. La información básica de seguridad todavía requiere presencia en el embalaje para asegurar el acceso sin dispositivos digitales.
La presión ambiental está reformulando las decisiones de envasado de formas que a veces entran en conflicto con los requisitos reglamentarios. El sector farmacéutico se enfrenta a exigencias para reducir los residuos y utilizar materiales sostenibles, objetivos que pueden chocar con reglamentos que especifican construcciones de envases particulares. La Unión Europea ha impulsado unos requisitos de envasado sostenibles especialmente agresivos que afectan al diseño y la producción de las etiquetas.
Las etiquetas inteligentes con chips NFC o tecnologías embebidas verifican la autenticidad de los productos mientras se realiza un seguimiento del uso y monitoreo de la exposición a la temperatura a lo largo de la distribución. Estas capacidades plantean nuevas cuestiones regulatorias sobre la privacidad y seguridad de los datos, así como hasta qué punto la funcionalidad de la etiqueta inteligente se extiende al territorio de los dispositivos médicos. La tecnología existe; el marco reglamentario sigue formándose.
Recursos para Profesionales de Etiquetado Físico
Asociaciones profesionales como la Regulatory Affairs Professionals Society (RAPS) y la Drug Information Association (DIA) lo conectan con personas que se enfrentan a desafíos similares de etiquetado a través de conferencias y grupos de trabajo. Usted tiene conocimiento de cómo los reguladores abordan los nuevos requisitos mientras ven cómo otras empresas resuelven los problemas con los que se enfrentan. RAPS también ofrece certificación de los asuntos regulatorios que cubre las competencias de etiquetado, mientras que los programas de capacitación especializados abordan lagunas específicas cuando los equipos necesitan ponerse al día en nuevas regulaciones o tecnologías.
El apoyo externo proviene de dos direcciones. Las empresas consultoras aportan experiencia regulatoria para navegar por complejas sumisiones, en particular cuando se trata de requisitos desconocidos o plazos ajustados. Los proveedores de tecnología ofrecen plataformas que manejan la gestión de versiones a través de los mercados al tiempo que coordinan cambios y aprobaciones. La elección entre ellos a menudo depende de si su reto es entender lo que los reguladores quieren o manejar la complejidad operativa de hacerlo.
Para los equipos que gestionan contenidos de alto nivel, la precisión no es opcional. La verificación de GlobalVison da a los equipos la confianza de que cada versión aprobada está lista para ser inspeccionada.
