
Quando um dispositivo médico estéril falha na cirurgia porque a sua embalagem não conseguiu manter a esterilidade durante o transporte, a segurança do paciente sofre primeiro. Retiradas dos produtos e reputação da empresa danificada seguem-se de perto.
Introdução à validação de pacotes
Validação de embalagens de dispositivos médicos prova que seus sistemas de embalagens protegem a esterilidade do dispositivo contra fabricação através de cuidados. As suas embalagens servem como um sistema estéril de barreiras que mantém a proteção microbiana durante a esterilização e distribuição, ajudando os dispositivos a permanecerem estéreis durante anos antes de chegarem aos pacientes.
ISO 11607 fornece uma estrutura internacional para validar esses sistemas. A Parte 1 define requisitos para materiais e sistemas de barreiras estéreis usados para embalar dispositivos médicos esterilizados terminalmente. A Parte 2 fornece requisitos de validação para a formação, eliminação e processos estéreis de sistema de barreiras. Juntos, asseguram que as embalagens são capazes de manter a esterilidade e provaram ser executadas em condições de produção no mundo real. A regulamentação do sistema de qualidade FDA (21 CFR 820) obriga a requisitos paralelos por meio de controles de design e de mandatos de validação de processo, tornar a validação uma necessidade de conformidade em vez de uma medida de qualidade opcional.
A validação é tipicamente liderada pela garantia de qualidade, mas o verdadeiro sucesso depende da coordenação entre departamentos. Muitos atrasos resultam da confusão sobre papéis ou lacunas nas expectativas. Quando a engenharia, a regulamentação e os grupos produtivos contribuem com a sua experiência em sincronização, a validação se torna muito mais eficiente e confiável.
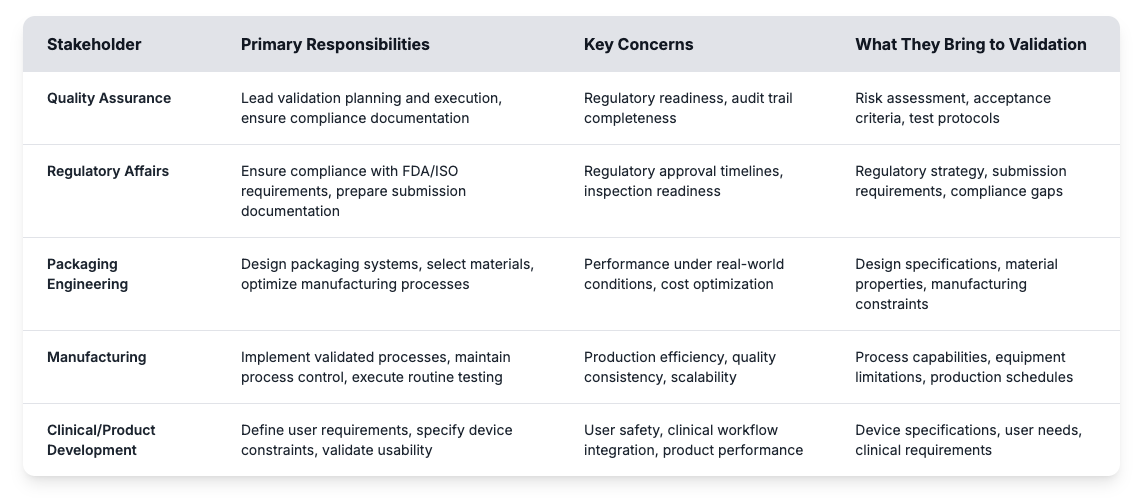
Conseguir esses papéis ajuda os fabricantes de dispositivos médicos a criarem programas de validação eficazes e evitar armadilhas comuns. A avaliação de risco identifica potenciais pontos de falha, enquanto os critérios de aceitação estabelecem metas mensuráveis para o desempenho das embalagens. Os protocolos de testes definem como os testes de validação provarão que os sistemas estéreis de barreiras cumprem os requisitos regulamentares ao longo de toda a vida útil pretendida. A construção de um programa de validação bem-sucedido começa com a compreensão desses requisitos fundamentais e o estabelecimento de processos claros que conectam as contribuições das partes interessadas a resultados mensuráveis.
Requisitos de Fundamento para validação de empacotamento
Todo programa de validação bem-sucedido começa com três elementos básicos de validação de embalagem que os reguladores esperam ver:
- Plano de validação abrangente: Seu plano de validação conecta decisões de design à documentação de conformidade. Ele define quem faz o quê, quando os testes acontecem, e como você documentará os resultados. Os reguladores analisam estes planos durante as inspecções para ver se a sua abordagem faz sentido ou se você é apenas caixas de verificação. Bons planos incluem protocolos claros com atribuições de responsabilidade e cronogramas realistas que respondem a possíveis atrasos.
- Structured risk assessment: Risk assessment helps you focus validation resources where they matter most. Você identificará cenários em que as embalagens possam comprometer a esterilidade ou a segurança do dispositivo - coisas como falhas de selo durante o envio, problemas materiais com seu método de esterilização, ou embalagens que são muito difíceis de serem abertas pelos usuários com segurança. Essa avaliação diz quais testes são críticos e quais são simpáticos.
- Ucritérios de aceitação cientificamente justificados: Critérios de aceitação definem o que "bom o suficiente" é para o desempenho de suas embalagens. Precisam de ser específicas e mensuráveis enquanto se mantêm vinculadas à segurança dos doentes e não à conveniência do fabrico. Em vez de critérios vagos como "aparência aceitável", você vai definir metas para a força das focas, taxas de vazamento e manutenção da esterilização que fazem sentido para o seu dispositivo específico e condições de uso.
Os alicerces fracos criam problemas de validação que multiplicam ao longo do progresso dos programas. Muitas vezes, as equipes descobrem lacunas demasiado tarde, ao corrigi-las significa começar com testes que deveriam ter sido incluídos desde o início. O efeito dominó é previsível: os testes dispersos conduzem a provas incompletas, o que cria questões regulamentares que atrasam as aprovações e aumentam os custos.
Os Quatro Pilares de Validação de Pacotes
ISO 11607 quebra a validação de embalagem em quatro pilares interconectados que trabalham juntos para provar que sua embalagem não irá falhar quando isso mais importa. Rather than treating these as separate checkboxes, successful teams understand how each pillar feeds into the next to build comprehensive evidence that packaging protects device sterility throughout the device lifecycle.

Você não pode provar um desempenho consistente em embalagens (PQ) sem saber se seu equipamento funciona de forma confiável (OQ). Você não pode confiar no desempenho de equipamento sem a instalação adequada (IQ). Qualificação de material (MQ) é executada juntamente com validação de equipamento para garantir que tudo funcione como um sistema completo de embalagens. Quando feita correctamente, esta abordagem sistemática constrói provas de validação passo a passo com cada pilar, reforçando os outros. Quando você ignora etapas, você cria problemas caros:
- Ignorar o IQ significa que você não sabe se os problemas do equipamento estão bagunçando seus resultados de testes posteriores.
- O OQ Fraco deixa você com parâmetros de processo que parecem bons em testes, mas que se desmoronam sob o estresse da produção.
- Qualificação material curto é como as equipes acabam com embalagens que falham durante o envio ou perdem esterilidade ao longo do tempo.
Pense em validação como construir uma casa - você não iria começar com o telhado antes de lançar uma base sólida. Os quatro pilares funcionam da mesma forma, com cada etapa a apoiar o que vem a seguir. Ignore o terreno e você vai gastar muito mais tempo e resolvendo problemas de dinheiro que poderiam ter sido evitados desde o início com uma abordagem sistemática.
Qualificação da instalação: Verificação de Equipamento e Calibração
Sem documentação de base, problemas de equipamento e falhas de processo se tornam impossíveis de serem separadas durante fases de validação posteriores. Qualificação de instalação (IQ) impede isso ao estabelecer exatamente com o que você está começando antes de iniciar qualquer teste de desempenho.
O núcleo produtivo a partir do IQ é a documentação do parâmetro base que captura o ponto de partida do seu equipamento. Você vai gravar:
- Intervalos de operação para parâmetros críticos como temperatura, pressão, velocidade e tempo de permanência
- Configurações padrão e controle de configurações do sistema
- Condições ambientais na área de embalagem
- Características do desempenho de equipamento em condições de ausência de carga
- Requisitos de manutenção preventiva e horários que mantêm o equipamento a funcionar de forma consistente
A verificação de equipamento e a calibração estabelecem a fundação para essas linhas de base. Você confirmará que todos os componentes correspondem às especificações de compra, que os utilitários atendem aos requisitos e que os sistemas de segurança funcionam corretamente. Para equipamento de vedação, você documenta recursos de temperatura e pressão juntamente com especificações do elemento de aquecimento. Os sistemas de controle são verificados para confirmar que eles podem manter os parâmetros necessários.
A calibração estende a base do IQ. Instrumentos que monitoram ou controlam o processo de embalagem - controladores de temperatura, medidores de pressão, sistemas de timing e ferramentas de teste - deve ter certificados rastreáveis até os padrões nacionais. Estes registram o desempenho do equipamento e demonstram o controle do processo durante a validação e produção rotineira.
Esse registro também se torna o ponto de referência para fases posteriores. Se a força da foca for alterada durante a qualificação de desempenho, os resultados poderão ser rastreados até a variação de equipamento documentada na QI. Sem este nível de documentação, torna-se difícil distinguir entre as questões relacionadas com o equipamento e as falhas do processo, levando a investigações dispendiosas que teriam evitado o trabalho de arranque completo.
Qualificação operacional: Testando todos os intervalos operacionais
Seu equipamento pode funcionar perfeitamente em condições ideais, mas dá resultados inconsistentes quando a produção fica confuso. Qualificação operacional (OQ) revela essas zonas de instabilidade antes que elas causem problemas a jusante. Ao testar parâmetros nos seus limites, as equipes podem ver onde o processo se mantém estável e onde a performance começa a falhar.
Seu objetivo é estabelecer parâmetros de processo que as equipes de produção usarão diariamente - os limites de operação, com pontos definidos normais mais intervalos de variação aceitáveis. Você encontrará intervalos onde o equipamento produz resultados confiáveis de forma consistente, com as melhores configurações que oferecem melhor desempenho e segurança que previnem problemas. Testar a funcionalidade de equipamento significa variar sistematicamente um parâmetro enquanto segura outros constantes, em seguida, monitorar os resultados de qualidade do pacote. Testar nas bordas ajuda a identificar onde o desempenho começa a falhar. Esta torna-se sua margem de segurança para a produção rotineira.
Registros OQ precisam ir além de listar o que o equipamento pode fazer. Deveriam igualmente captar as condições que mantêm os resultados coerentes. Por exemplo, uma alta humidade pode interferir na vedação, mudanças de temperatura podem causar derivação em parâmetros críticos, e máquinas próximas podem introduzir vibrações que reduzem a precisão.
Este teste captura combinações de parâmetros que criariam problemas na produção antes de aparecerem. Seu equipamento pode ser executado em certas configurações, mas dá resultados inconsistentes que causam dores de cabeça a jusante. OQ identifica essas zonas de problema para que equipes de produção possam ser claras com as gamas que você estabelecer aqui se tornando a base para treinamentos de operadores e processar alarmes que continuam a fabricar sem problemas.
Qualificação do desempenho: Protestando Desempenho Consistente de Empacotamento
Qualificação de desempenho (PQ) demonstra que o seu processo completo de embalagem produz consistentemente pacotes que cumprem todos os critérios de aceitação em condições reais de produção. É aqui que a validação se move de provar capacidade para provar confiabilidade.
A realidade é que o vosso processo de embalagem precisa funcionar quando tudo não é perfeito. Configurações de equipamentos derivam um pouco, os operadores mudam entre turnos e os lotes de material variam de qualidade. As condições ambientais flutuam durante o dia. Testes do QG mostram que seu processo ainda produz pacotes aceitáveis apesar dessas variações normais. Os testes de cenários de pior qualidade levam isso mais longe, empurrando seu processo validado para limites realistas. Os testes de esforço devem reflectir as realidades da produção e não apenas as condições ideais. Isso pode significar funcionar a velocidades máximas de linha enquanto os operadores pressionam para cumprir prazos, trabalhando em controles ambientais reduzidos se os sistemas HVAC não estiverem funcionando bem, ou a qualidade de teste no final de uma mudança quando o cansaço puder influenciar os resultados.
Executar testes de validação em condições reais de produção significa utilizar os mesmos materiais, configurações de equipamento e condições ambientais que as equipes de produção enfrentam diariamente. Capturar variação do processo natural ao longo do tempo, operadores, lotes materiais e equipamento estados mais do que testar em condições artificialmente controladas que não representam a realidade. Planejamento de estatísticas define a quantidade de testes necessários para provar um desempenho consistente. Um plano de amostragem bem projetado abrange várias execuções e operadores, garantindo que os resultados refletam a capacidade real do processo em vez de o acaso. Essa evidência conduz então à monitorização do processo e sustenta o desempenho validado durante a produção quotidiana.
Qualificação dos Materiais e Design: Compatibilidade e Proteção
Qualificação do material (MQ) mostra que os materiais de embalagem do dispositivo podem ser executados como previsto através do ciclo de vida completo do dispositivo. Para dispositivos médicos esterilizados terminalmente, isto significa selecionar embalagens primárias que suportem o método de esterilização escolhido e continuam a proteger a esterilidade durante a distribuição e o armazenamento. A validação do sistema de empacotamento em seguida confirma que os materiais, o design e a função de vedação juntos como uma barreira estéril ao longo da vida pretendida do dispositivo.
Ao selecionar materiais, as equipes devem avaliar três fatores:
- O método de esterilização que será usado
- O ambiente de armazenamento que o pacote deve resistir
- Os requisitos do usuário para abertura segura e acesso ao dispositivo
Cada método de esterilização apresenta desafios únicos. A esterilização de óxido de Ethyleno requer tipicamente materiais que resistem à degradação química, ao mesmo tempo que permitem a penetração e evacuação do gás para uma esterilização e aeronáutica eficazes. A esterilização Gama expõe as embalagens a radiações que podem enfraquecer a força e a flexibilidade. A esterilização do Steam combina alta temperatura com umidade que pode comprometer as propriedades de barreiras. Se esses riscos não forem abordados cedo, a barreira estéril pode falhar no uso do mundo real, colocando em risco a conformidade e a segurança dos pacientes.
Testes de compatibilidade de materiais avaliam como a sua embalagem interage com os componentes do dispositivo ao longo de toda a vida na prateleira pretendida, mas também precisa abordar a usabilidade prática. Os materiais devem também manter as suas propriedades de barreira microbiana para garantir que as barreiras estéreis continuem a bloquear os contaminantes ao longo da vida na prateleira pretendida. Ao mesmo tempo, A força das peles tem de ser suficientemente forte para manter a integridade das focas durante a distribuição, mas suficientemente gentil para que os prestadores de cuidados de saúde abram de forma aseptica sem lágrimas. Você realizará estudos acelerados que simulam anos de armazenamento em semanas, combinado com a idade em tempo real em condições de armazenamento, para confirmar o atraso das suas previsões.
Protocolos de Teste Material:
| Tipo de teste | Avaliações de chaves | Objetivo |
|---|---|---|
| Física | Força das focas, teste explosivo para resistência à pressão, teste de tintura para detecção de vazamento | Integridade mecânica |
| Química | Extrtabelas, leacaso, esterilizantes residuais | Segurança dos materiais |
| Biológico | Manutenção de esterilização, biocompatibilidade | Segurança do paciente |
Os protocolos iniciais de teste ajudam a estabelecer o desempenho do material de base antes do início de estudos de validação completos. Concentre-se em projetar protocolos de teste que comportam os materiais de estresse da maneira como eles realmente serão estressados no uso do mundo real, em vez de atender a especificações arbitrárias de teste que não se relacionam com requisitos de desempenho reais. Características de abertura devem permitir um acesso limpo e controlado que não comprometa a esterilidade ou a dispersão no campo estéril, o que significa que o seu teste precisa simular condições reais de uso clínico em vez de ambientes laboratoriais ideais.
Distribuição e teste do trânsito: Simulando envio mundial real
As embalagens têm de resistir a toda a viagem, desde o piso de produção até ao berço do doente. Testes de distribuição recriam as tensões de transporte e manuseio para que as equipes possam confirmar que as barreiras estéreis permanecem intactas e funcionais em toda a cadeia de suprimentos.
ASTM D4169 e protocolos ISTA esboça como simular ambientes de distribuição, mas você precisa adaptá-los às suas condições de envio. Em vez de controlos isolados, o teste deveria reflectir toda a viagem: um pacote pode ser descartado, atormentado por vibrações de transporte ou esmagado sob cargas de armazéns. No caminho, a orientação muda, a resolução repetida e a alteração das condições climáticas testam a força da barreira estéril. Executar essas tensões em sequência dá uma imagem mais verdadeira de como a embalagem funciona na distribuição no mundo real.
As embalagens passam por várias verificações após testes de distribuição. A inspecção visual procura por danos de superfície, enquanto os testes de força confirmam que os encerramentos permanecem intactos. Métodos mais sensíveis, como testes de bolha, descubra micro-fugas em barreiras estéreis que não podem ser vistas pelos olhos. A etapa final é o teste de integridade, a prova de que a barreira microbiana continua a funcionar mesmo depois de a navegação simulada - e esses resultados só são importantes se os testes reflectirem as condições reais de navegação. Embalagens que aprovam protocolos genéricos mas falham no terreno levam a que se recordem e prejudicam a reputação da empresa. Mais importante ainda, colocam os doentes em risco.

Estudos em Tempo Real e Acelerado: Reivindicações de Vida Provisória
A Shelf-life afirma ou invoca provas de que as embalagens preservam a sua barreira estéril enquanto os dispositivos permanecerem no mercado. A idade acelerada dá aos fabricantes uma visão precoce do desempenho das embalagens, utilizando a temperatura elevada e a umidade para simular anos de armazenamento em apenas semanas. ASTM F1980 (versão atual) traça a abordagem mas a verdadeira dificuldade é conceber condições que espelhem ambientes reais sem introduzir modos de fracasso que nunca viriam a acontecer na prática. Quando bem concebidos, estes estudos produzem dados preliminares que apoiam pretensões iniciais de estadia e ajudam os produtos a avançar, enquanto ainda estão em curso estudos a longo prazo.
Tempo real ele completa a imagem. Os pacotes de produção final são armazenados em condições verdadeiras e monitorados por desempenho ao longo de meses ou anos. Esta prova demonstra como as embalagens resistem à realidade quotidiana do armazenamento e da distribuição, proporcionando expectativas aos reguladores de dados confirmados. Equipes que iniciam estudos em tempo real evitam lacunas de dados que podem atrasar as aprovações ou renovações mais tarde.
As determinações mais confiáveis da estante vêm da combinação de projeções aceleradas com confirmação em tempo real. Análise estatística mostra quando propriedades de embalagem caem abaixo de seus objetivos e lhe dão intervalos de confiança para reivindicações de vida na prateleira. O planeamento fraco, no entanto, pode minar todo o esforço - testes de protótipos em vez de runas de produção, selecionando condições de armazenamento que não refletem a realidade ou subestimar tamanhos amostrais toda a credibilidade de erosão.
Com a abordagem correta, estudos em idade comprovam que as embalagens protegerão os doentes ao longo do período de vida pretendido, dando aos fabricantes e às entidades reguladoras a confiança de que as alegações se manterão ao longo do tempo. Estas provas vão além dos requisitos regulamentares para demonstrar o desempenho real do mundo que é importante para a segurança dos doentes.
Avaliação de usabilidade e fatores humanos: validação de interação do usuário
Um sistema de embalagem pode atender todos os padrões técnicos e ainda falhar se não puder ser aberto com segurança nas mãos de um médico. A validação de usabilidade garante que os sistemas de barreiras estéreis não retenham apenas no laboratório, mas também no momento real em que são abertos os dispositivos de assistência aos doentes. Tanto a FDA como a ISO 11607 ligam a usabilidade directa à gestão de riscos, reconhecendo que as embalagens que comprometem a técnica da aseptic são efectivamente um sistema falhado.
A central focus is aseptic presentation testing - proof that the sterile barrier can be opened safely in practice. Os sistemas de encerramento e as focas de embalagem têm de resistir a um fracasso prematuro, mas sem problemas em condições clínicas. A integridade das focas é essencial. Os pacotes precisam de descascar sem rasgar, e o desenho deve manter as partículas ou os detritos fora do campo estéril. Um problema frequente é a foca excessivamente engenhosa - suficientemente forte para cumprir as metas dos testes, mas tão difícil de abrir que a técnica aseptica se torna impraticável. Teste de uso simulado ajuda a supervisionar esses problemas antes de os produtos chegarem ao mercado.
Na prática, os testes envolvem frequentemente enfermeiros ou técnicos cirúrgicos que abrem pacotes em condições estéreis, enquanto os observadores tomam nota de quaisquer interrupções no técnico. O tipo de luva importa: nitrilo e latex fornecem empunhadura e sensação diferentes. Outros fatores do mundo real - como a iluminação Ou, a pressão do tempo durante os procedimentos, ou como o próprio pacote coopera bem - também afecta a capacidade de manter a esterilidade. Mesmo detalhes como guias de casca podem fazer a diferença: fácil de se gerenciar com as mãos vazias, mas quase impossível com luvas. A força excessiva da foca representa o mesmo risco, forçando as voltas que minam todo o propósito das embalagens esterilizadas. Um selo que não pode ser aberto aseticamente não apenas frustra os usuários, ele compromete diretamente a esterilidade no ponto de cuidados.
Igualmente importante é compreender como as pessoas interagem com o pacote na prática. Estudos de usabilidade observam os prestadores de cuidados de saúde que estão a abrir dispositivos em condições realistas, surjam momentos de hesitação juntamente com variações técnicas ou riscos de contaminação potencial. Essas observações revelam problemas que os testes materiais falham - selos que exigem força excessiva ou embalagens que lágrimas de forma imprevisível quando os usuários precisam de acesso rápido. A falta de clareza da abertura cria confusão que compromete a técnica estéril durante os procedimentos reais.
Finalmente, a validação deve ter em conta requisitos de treinamento. Se o uso seguro depender de instruções complexas, o risco de erro aumenta. Avaliar o treinamento precisa de ajuda a confirmar que a embalagem é intuitiva para os usuários pretendidos. Todas as instruções essenciais para um tratamento seguro devem ser documentadas com clareza. As equipes que tomam este passo à frente estão melhor posicionadas para evitar redesenhos dispendiosos ou reclamações inesperadas após o lançamento.
Quando a usabilidade é incorporada à validação desde o início, os fabricantes obtêm mais do que evidências de conformidade. Proporcionam embalagens que funcionam tanto para os pacientes como para os médicos, garantindo ao mesmo tempo aos reguladores que executa onde mais importa.
Qualificação do Desempenho do Processo: Validando consistência de Fabricação
Até mesmo o pacote mais cuidadosamente projetado falha se o processo de fabrico não puder entregar o mesmo resultado toda vez. A Qualificação de Desempenho do Processo (PPQ) prova que as embalagens não são apenas bem-sucedidas no laboratório ou durante testes de validação, mas permanecem consistentes em toda a escala de produção. Este é o ponto em que tanto os reguladores como as equipas de qualidade internas precisam de prova de que os sistemas de embalagens podem resistir à produção mundial.
Um QP forte examina toda a sequência de produção, incluindo processos de montagem. Analisa a forma como os materiais são armazenados, como os operadores lidam com os componentes e se os controlos ambientais mantêm condições estáveis. Qualquer fraqueza nessas áreas pode erodir o desempenho das embalagens, mesmo quando as configurações do equipamento estão discadas.
Confiabilidade vem da repetibilidade. É por isso que o PPQ exige que várias execuções de produção comprovem que o processo oferece resultados consistentes em diferentes lotes. Equipes normalmente validam três lotes consecutivos para mostrar que o desempenho é consistente ao longo de diferentes execuções de produção, embora os requisitos possam variar consoante o risco do produto e a orientação regulamentar. Seguidamente, o acompanhamento sustenta essa garantia, apanhando a deriva antes de se transformar em questões de qualidade.
O pagamento estende-se muito para além do cumprimento. Um PPQ bem executado oferece benefícios que atingem operações diárias. Torna a produção mais previsível enquanto acelera as decisões de libertação. Mais importante ainda, cria confiança junto dos reguladores quanto ao facto de o sistema estar sob controlo.
Revalidação de Requisitos e alteração de Gestão
A mudança é inevitável na produção e todas as mudanças levantam a questão de saber se o seu sistema de embalagens ainda faz o seu trabalho. A revalidação fornece a evidência de que sim, impedindo que pequenos ajustes criem riscos grandes. Os reguladores vêem a mudança descontrolada como uma bandeira vermelha, e os fabricantes também devem fazê-lo. Gatilhos comuns incluem:
- Atualizações de equipamentos, tais como novos sistemas de vedação ou inspecção
- A esterilização muda, se uma mudança no método ou ajustes de parâmetro
- Modificações de material, de mudar de fornecedores para selecionar substratos diferentes
- Processar mudanças, mesmo as menores que alteram condições de operação
The scope of revalidation depends on risk and documented validation requirements. Any change to a process raises the question of sterility. As equipes podem descobrir que um pequeno ajuste afeta a integridade das focas. Noutros casos, a compatibilidade material tem de ser reavaliada. Por vezes, a preocupação é saber se as propriedades das barreiras microbianas continuarão a existir. Seja qual for o gatilho, os testes de acompanhamento são a prova de que as embalagens continuam a proteger como pretendido. Análises regulares também são esperadas, uma vez que o equipamento usa ao longo do tempo. As deslocações nos fornecedores ou a deriva gradual do processo podem ter o mesmo impacto, e cada um deve ser monitorizado.
Uma boa documentação reúne tudo isto para que os reguladores de provas possam confiar. Deve explicar o raciocínio por trás de cada teste, e não apenas capturar esse teste. Limpar registros fornecem mais do que um rastro de papel. Dão confiança aos reguladores, ajudam as equipes a gerir a mudança de forma proactiva e, acima de tudo, mostram que as embalagens continuam a proteger os doentes muito para além do primeiro relatório de validação.
Desafios comuns e soluções eficazes
A validação pode tropeçar por razões familiares. Os tamanhos de amostragem são muitas vezes subestimados, os testes com pior caso são ignorados e as notas de documentação até que deixe de refletir o que foi feito. As equipas também passam horas comparando manualmente documentos e fingindo que perceberam se perceberam todas as mudanças críticas. Cada um destes passos errados pode se sentir pequeno no momento, mas em conjunto geram retrabalhos dispendiosos e contam com conclusões regulamentares que prejudicam a credibilidade muito depois de a auditoria terminar. O estrangulamento da revisão manual é particularmente problemático porque encolhe profissionais de qualidade experientes em comparações tediosas em vez de aplicar os seus conhecimentos na avaliação dos riscos e na tomada de decisões estratégicas.
A abordagem mais forte consiste em construir a validação como actividade estratégica. Critérios claros de aceitação definidos desde o início ajudam a evitar desacordos tardios, especialmente quando a qualidade, a engenharia e as equipes reguladoras se alinham cedo no processo. Quando a validação corre ao longo de todo o desenvolvimento, em vez de ser tratada como um passo final, as lacunas tornam-se mais fáceis de identificar enquanto as revisões regulamentares evoluem mais facilmente.
Trabalhar de forma eficiente significa fazer esforços onde mais conta, e não apenas fazer cortes. A amostragem baseada nos riscos concentra-se nos recursos onde o fracasso seria mais importante. Os planos de testes podem ser concebidos para servir mais de um requisito por vez, reduzindo o despedimento sem reduzir o rigor. Utilizadas desta forma, as melhores práticas poupam tempo ao mesmo tempo que reforçam a base de provas, garantindo que as embalagens ganham confiança no regulador e continua a proteger os pacientes em uso.
Documentação e envio regulatório
Para os reguladores, se não estiver documentado, isso não aconteceu. Os fabricantes enfrentam a mesma realidade: os registos incompletos podem atrasar as aprovações, ao mesmo tempo que desencadeiam conclusões de inspecções que enfraquecem a confiança no sistema de qualidade.
Documentação forte fecha essas lacunas. Um registro completo mostra como o teste foi planejado e executado ao conectar os resultados de volta aos critérios de aceitação. Esta documentação demonstra a conformidade da regulamentação. Certificados de validação e arquivos técnicos formalizam depois essa prova, confirmando que os sistemas de embalagens cumprem as expectativas ISO 11607 e FDA. Mais do que um binder de relatórios, a documentação eficaz junta protocolos, dados e avaliações de risco numa história de conformidade controlada e pronta para inspeção. Quando os reguladores perguntam, os fabricantes têm de mostrar não só o que foi testado, mas também porquê, como e com que resultado.
Embora a documentação completa não satisfaça apenas os reguladores; ela mantém os dispositivos movendo-se para o mercado sem interrupções dispendiosas. Quando os registos estiverem completos e estiverem prontos para inspecção, os fabricantes podem defender o seu processo com confiança e os doentes recebem embalagens que os protejam ao longo do ciclo de vida do dispositivo.
Garantir sucesso do sistema de pacotes
A validação de embalagens prova que os sistemas estéreis de barreiras podem resistir à jornada desde o andar da fábrica até ao ponto de cuidado. Com base num planeamento cuidadoso e apoiado por ensaios e documentação sólidos, a validação dá aos fabricantes confiança na realização das suas embalagens no terreno e satisfaz os reguladores. Tratar a validação como uma responsabilidade do ciclo de vida, não como um exercício único, evitar falhas dispendiosas e reforçar a confiança tanto entre equipas de qualidade como entre reguladores.
A paisagem está evoluindo. Plataformas de validação digital e ligações mais apertadas à gestão do risco estão a reformular as melhores práticas, enquanto os reguladores se movem para um maior alinhamento global. O que não mudará é o objectivo: assegurar que as embalagens mantenham a esterilidade, para que os clínicos possam confiar em todos os dispositivos e para que os doentes recebam a protecção que merecem. Olhando para o futuro, uma integração mais estreita da validação de embalagens com validação de esterilização irá moldar a prática da indústria, garantindo que as alegações de esterilidade sejam apoiadas interminavelmente.
GlobalVision’s Verify accelerates packaging validation by automating document and artwork inspection, giving teams compliance confidence without slowing them down.
