
Cuando un dispositivo médico estéril falla en la cirugía porque su embalaje no podía mantener la esterilidad durante el envío, la seguridad del paciente sufre primero. Las retiradas de productos y la reputación dañada de la empresa siguen de cerca.
Introducción a la validación del empaquetado
La validación del embalaje de dispositivos médicos demuestra que sus sistemas de embalaje protegen la esterilidad de los dispositivos de fabricación a través del punto de atención. Su embalaje sirve como una barrera estéril que mantiene la protección microbiana a través de la esterilización y distribución, ayudando a que los dispositivos permanezcan estériles durante años antes de llegar a los pacientes.
ISO 11607 proporciona el marco internacional para validar estos sistemas. La Parte 1 establece requisitos para materiales y sistemas de barrera estériles utilizados para empaquetar dispositivos médicos esterilizados terminalmente. La Parte 2 proporciona requisitos de validación para los procesos de formación, sellado y de sistemas de barrera estéril. Juntos, aseguran que los envases sean capaces de mantener la esterilidad y demuestren su desempeño en condiciones de fabricación reales. Norma del Sistema de Calidad de la FDA (21 CFR 820) impone requisitos paralelos a través de controles de diseño y obligatorios de validación de procesos, hacer de la validación una necesidad de cumplimiento en lugar de una medida de calidad opcional.
La validación está típicamente dirigida por la garantía de calidad, pero el éxito real depende de la coordinación entre departamentos. Muchos retrasos provienen de la confusión sobre los papeles o lagunas en las expectativas. Cuando los grupos de ingeniería, regulación y manufactura contribuyen con su experiencia en la sincronización, la validación se vuelve mucho más eficiente y fiable.
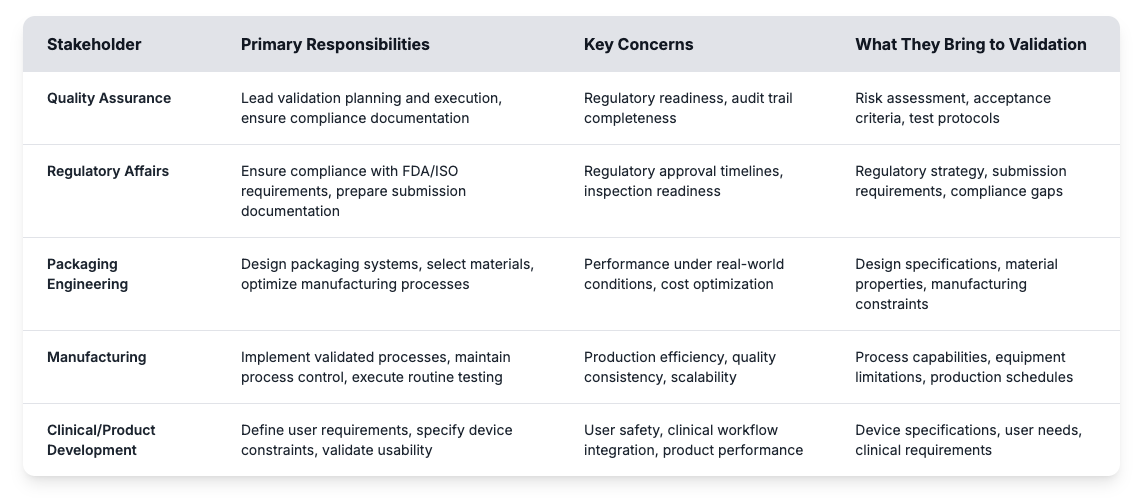
Lograr estos roles correctamente ayuda a los fabricantes de dispositivos médicos a crear programas de validación efectivos y a evitar las caídas comunes. La evaluación de riesgos identifica los posibles puntos de fracaso, mientras que los criterios de aceptación establecen objetivos asequibles para el desempeño de los envases. Los protocolos de prueba definen cómo las pruebas de validación demostrarán que los sistemas de barrera estériles cumplen los requisitos regulatorios a lo largo de la vida útil de la protección prevista. La creación de un programa de validación exitoso comienza con la comprensión de estos requerimientos fundamentales y el establecimiento de procesos claros que conecten la entrada de las partes interesadas con resultados que se puedan alcanzar.
Requisitos fundacionales para la validación del empaquetado
Cada programa de validación exitoso comienza con tres elementos básicos de validación del embalaje que los reguladores esperan ver:
- Comprehensive validation plan: Your validation plan connects design decisions to compliance documentation. Define quién hace qué, cuando pasa la prueba, y cómo resultará el documento. Los reguladores examinan estos planes durante las inspecciones para ver si su enfoque tiene sentido o si sólo está marcando casillas. Los buenos planes incluyen protocolos claros con asignaciones de responsabilidad además de cronogramas realistas que explican los posibles retrasos.
- Evaluación de riesgos estructurada: La evaluación de riesgos le ayuda a centrar los recursos de validación donde más importan. Usted identificará escenarios donde el embalaje podría comprometer la esterilidad o seguridad del dispositivo - cosas como fallos de sellado durante el envío, problemas materiales con su método de esterilización o empaque que es demasiado difícil para que los usuarios puedan abrirse de forma segura. Esta evaluación le indica qué pruebas son críticas y cuáles son agradables.
- Scientifically justified acceptance criteria: Acceptance criteria define what "good enough" looks like for your packaging performance. Necesitan ser específicos y comprensibles mientras permanecen vinculados a la seguridad del paciente en lugar de a la conveniencia de fabricar. En lugar de criterios vagos como "apariencia aceptable", establecerás objetivos de fuerza sellada, tasas de fugas, y mantenimiento de la esterilidad que tienen sentido para su dispositivo específico y condiciones de uso.
Las fundaciones débiles crean problemas de validación que se multiplican a medida que progresan los programas. Los equipos a menudo descubren huecos demasiado tarde, cuando arreglarlos significa comenzar con pruebas que deberían haber sido incluidas desde el principio. El efecto dominó es predecible: las pruebas dispersas conducen a pruebas incompletas, lo que crea cuestiones reglamentarias que retrasan las aprobaciones y aumentan los costos.
Los cuatro pilares de la validación del embalaje
ISO 11607 divide la validación de empaques en cuatro pilares interconectados que trabajan juntos para demostrar que tu empaquetado no fallará cuando más importe. Rather than treating these as separate checkboxes, successful teams understand how each pillar feeds into the next to build comprehensive evidence that packaging protects device sterility throughout the device lifecycle.

No puede demostrar un rendimiento consistente del embalaje (PQ) sin saber que su equipo funciona de forma fiable (OQ). No puede confiar en el rendimiento del equipo sin una instalación adecuada (IQ). La cualificación de los materiales (MQ) va acompañada de la validación de los equipos para asegurar que todo funcione conjuntamente como un sistema de embalaje completo. Cuando se hace correctamente, este enfoque sistemático crea pruebas de validación paso a paso con cada pilar que refuerza los demás. Cuando omites los pasos, creas problemas caros:
- Omitir el IQ significa que no sabe si los problemas de equipamiento están alterando sus resultados de prueba posteriores.
- Débil OQ le deja con parámetros de proceso que se ven bien en las pruebas pero que se descomponen bajo el estrés de producción.
- La calificación de material de corta duración es la forma en que los equipos terminan con un envase que falla durante el envío o pierde esterilidad con el tiempo.
Piensa en la validación como construir una casa - no empezarías con el techo antes de sentar una base sólida. Los cuatro pilares funcionan de la misma manera, y cada etapa apoya lo que viene a continuación. Omita el trabajo preliminar y pasará mucho más tiempo y dinero resolviendo problemas que podrían haberse evitado con un enfoque sistemático desde el principio.
Calibración de Instalación: Verificación y Calibración de Equipos
Sin documentación de base sólida, los problemas de equipo y los fallos de proceso se vuelven imposibles de separar durante las etapas posteriores de validación. La cualificación de la instalación (IQ) evita esto estableciendo exactamente con qué está empezando antes de que comience cualquier prueba de rendimiento.
El núcleo entregable desde IQ es la documentación de parámetros de referencia que captura el punto de partida de su equipo. Registrarás:
- Rango operativo para parámetros críticos como temperatura, presión, velocidad y tiempo de morada
- Configuración predeterminada y configuraciones del sistema de control
- Condiciones medioambientales en el área de embalaje
- Características de rendimiento del equipo en condiciones de no carga
- Requisitos de mantenimiento preventivo y programas que mantienen el funcionamiento del equipo consistente
La verificación y la cualificación de los equipos establecen los cimientos para estas líneas de referencia. Confirmará que todos los componentes coinciden con las especificaciones de compra, las utilidades cumplen los requisitos y los sistemas de seguridad funcionan correctamente. Para los equipos de sellado, documentará las capacidades de temperatura y presión junto con las especificaciones del elemento de calefacción. Los sistemas de control se verifican para confirmar que pueden mantener los parámetros requeridos.
La calibración extiende la base del IQ. Instrumentos que monitorean o controlan el proceso de embalaje - controladores de temperatura, gaugas de presión, sistemas de temporización, y herramientas de prueba - deben tener certificados que se puedan rastrear hasta las normas nacionales. Estos registros registran el rendimiento de los equipos y demuestran el control del proceso durante la validación y la producción de rutina.
Ese conjunto de registros también se convierte en el punto de referencia para las etapas posteriores. Si la fuerza del sello se desliza durante la calificación de rendimiento, los resultados se pueden rastrear hasta la variación del equipo documentada en el IC. Sin este nivel de documentación, resulta difícil distinguir entre los problemas de los equipos y los fallos de los procesos, lo que conduciría a costosas investigaciones que los trabajos preliminares habrían evitado.
Cualificación Operacional: Pruebas a lo largo de los rangos operacionales
Su equipo puede funcionar perfectamente en condiciones ideales, pero dar resultados inconsistentes cuando la producción se torna complicada. La cualificación operacional (OQ) revela estas zonas de problemas antes de que causen problemas aguas abajo. Mediante la prueba de parámetros en sus límites, los equipos pueden ver donde el proceso se mantiene estable y donde el rendimiento comienza a descomponer.
Su objetivo es establecer parámetros de proceso que los equipos de producción usarán diariamente: los límites de funcionamiento con puntos normales más rangos de variación aceptables. Encontrará rangos en los que el equipo produce resultados fiables consistentemente, con ajustes óptimos que proporcionan el mejor rendimiento y márgenes de seguridad que evitan problemas. Probar la funcionalidad del equipo significa variar sistemáticamente un parámetro mientras se mantienen sostenidas otras constantes y luego monitorear las salidas de la calidad del paquete. Las pruebas en los bordes ayudan a identificar dónde el rendimiento empieza a desplomarse. Esto se convierte en su margen de seguridad para la producción de rutina.
Los registros OQ tienen que ir más allá de la lista de lo que el equipo puede hacer. También deben capturar las condiciones que mantienen consistentes los resultados. Por ejemplo, la alta humedad puede interferir con el sellado, los cambios de temperatura pueden provocar la deriva en parámetros críticos y la maquinaria cercana podría introducir vibraciones que reducen la precisión.
Esta prueba captura combinaciones de parámetros que crearían problemas en la producción antes de que aparezcan. Tu equipo puede funcionar en ciertos ajustes, pero da resultados inconsistentes que causan dolores de cabeza aguas abajo. OQ identifica estas zonas de problemas para que los equipos de producción puedan ser claros, con las gamas que usted establece aquí se convierte en la base para la formación del operador y las alarmas de proceso que siguen fabricando sin problemas.
Cualificación de Rendimiento: Prueba Consistente Desempeño del Embalaje
La cualificación de rendimiento (PQ) demuestra que su proceso completo de embalaje produce consistentemente paquetes que cumplen con todos los criterios de aceptación bajo condiciones reales de producción. Aquí es donde la validación pasa de la demostración de capacidad a la demostración de fiabilidad.
La realidad es que su proceso de embalaje necesita funcionar cuando todo no es perfecto. Los ajustes de equipamiento van a la deriva, los operadores cambian entre los turnos y los lotes de material varían en calidad. Las condiciones ambientales se fluctuan a lo largo del día. PQ testing muestra que su proceso todavía produce paquetes aceptables a pesar de estas variaciones normales. En el peor de los casos, la prueba de escenarios lleva esto más lejos al empujar su proceso validado a límites realistas. Las pruebas de resistencia deben reflejar las realidades de la producción, no sólo las condiciones ideales. Esto puede significar correr a velocidades máximas de línea mientras que los operadores empujan hacia el cumplimiento de los plazos, trabajar bajo controles ambientales reducidos si los sistemas HVAC no están funcionando bien, o probar la calidad al final de un cambio cuando la fatiga puede influir en los resultados.
Ejecutar pruebas de validación bajo condiciones de producción reales significa utilizar los mismos materiales, configuraciones de equipos y condiciones ambientales que los equipos de fabricación enfrentan diariamente. Capturar la variación natural de los procesos a través del tiempo, los operadores, las cantidades de materiales y los estados del equipo importa más que las pruebas bajo condiciones controladas artísticamente que no representan la realidad. La planificación estática establece la cantidad de pruebas necesarias para demostrar un desempeño consistente. Un plan de muestreo bien diseñado cubre múltiples operaciones y operadores, asegurando que los resultados reflejen la capacidad real del proceso en lugar de la casualidad. Esa evidencia impulsa el monitoreo del proceso y sostiene el desempeño validado durante la fabricación diaria.
Cualificación de materiales y diseño: Garantizar compatibilidad y protección
La cualificación del material (MQ) muestra que los materiales de embalaje de los dispositivos pueden funcionar como se pretende en todo el ciclo de vida del dispositivo. Para los dispositivos médicos esterilizados terminalmente, esto significa seleccionar envases primarios que resistan el método de esterilización elegido y que siguen protegiendo la esterilidad durante la distribución y el almacenamiento. La validación del sistema de embalaje confirma entonces que los materiales, el diseño y el sellado funcionan juntos como una barrera estéril a lo largo de la vida útil del dispositivo.
Al seleccionar materiales, los equipos deben evaluar tres factores:
- El método de esterilización que se utilizará
- El entorno de almacenamiento que el paquete debe soportar
- Requisitos de usuario para abrir y acceder a dispositivos de forma segura
Cada método de esterilización presenta desafíos únicos. La esterilización del óxido de etileno requiere típicamente materiales que resistan la degradación química al tiempo que permiten la penetración y evacuación de gas para una esterilización y aireación efectivas. La esterilización de gamma expone los envases a la radiación que puede debilitar la fuerza y la flexibilidad. La esterilización a vapor combina alta temperatura con humedad que puede comprometer las propiedades de barrera. Si estos riesgos no se abordan pronto, la barrera estéril puede fallar en el uso del mundo real, lo que pone en riesgo tanto el cumplimiento como la seguridad de los pacientes.
La prueba de compatibilidad de materiales evalúa cómo su embalaje interactúa con los componentes del dispositivo a lo largo de toda la vida útil prevista para la protección, pero también necesita abordar la usabilidad práctica. Los materiales también deben mantener sus propiedades de barrera microbiana para asegurar que las barreras estériles sigan bloqueando a los contaminantes a lo largo de toda la vida útil de la protección prevista. Al mismo tiempo, la fuerza de la piel debe ser lo suficientemente fuerte como para mantener la integridad del sello durante la distribución, pero lo suficientemente suave como para que los proveedores de atención médica se abran asépticamente sin desgarrar. Ejecutará estudios de envejecimiento acelerado que simulan años de almacenamiento en semanas, combinado con el envejecimiento en tiempo real en condiciones de almacenamiento para confirmar sus predicciones se sostienen.
Protocolos de prueba de materiales:
| Tipo de prueba | Evaluaciones clave | Propósito |
|---|---|---|
| Física | Sello de fuerza, ráfaga de resistencia a la presión, prueba de tinte para detección de fugas | Integridad mecánica |
| Química | Extractables, lixiviables, esterilantes residuales | Seguridad material |
| Biológico | Mantenimiento de la esterilidad, biocompatibilidad | Seguridad del paciente |
Los protocolos de pruebas iniciales ayudan a establecer el desempeño del material de referencia antes de que comiencen los estudios de validación completos. Concéntrese en el diseño de protocolos de prueba que estresen los materiales de la manera en que realmente se estrenarán en el uso del mundo real en lugar de cumplir con especificaciones de prueba arbitrarias que no se relacionan con los requisitos de rendimiento reales. Las características de apertura deberían permitir un acceso limpio y controlado que no comprometa la esterilidad o las partículas dispersas en el campo estéril, lo que significa que sus pruebas necesitan simular las condiciones reales de uso clínico en lugar de ambientes de laboratorio ideales.
Distribución y Prueba de Tránsito: Simulación de Envío Real-Mundo
El embalaje debe soportar el viaje completo desde el suelo de fabricación hasta la cama del paciente. Las pruebas de distribución recrea el estrés del envío y manipulación para que los equipos puedan confirmar que las barreras estériles permanecen intactas y funcionales a lo largo de la cadena de suministro.
ASTM D4169 and ISTA protocols outline how to simulate distribution environments, but you need to tailor them to your actual shipping conditions. En lugar de comprobaciones aisladas, las pruebas deberían reflejar el viaje completo: un paquete puede ser soltado, atacado por vibraciones de transporte o aplastado bajo cargas de almacén. A lo largo del camino, la orientación cambia, el manejo repetido y el cambio de condiciones climáticas ponen a prueba la fuerza de la barrera estéril. Ejecutar estos estrés en secuencia da una imagen más veraz de cómo el empaquetado funciona en la distribución del mundo real.
Los paquetes pasan por varias comprobaciones después de las pruebas de distribución. La inspección visual busca daños superficiales, mientras que la prueba de resistencia confirma que los cierres permanecen intactos. Métodos más sensibles, como pruebas de burbujas, descubren fugas de microbios en barreras estériles que el ojo no puede ver. El paso final es la prueba de integridad, la prueba de la barrera microbiana todavía se realiza incluso después de un envío simulado - y esos resultados sólo importan si las pruebas reflejan las condiciones reales de envío. Los paquetes que pasan los protocolos genéricos pero que fallan en el campo conducen a retiros a la vez que dañan la reputación de la empresa. Lo más importante es que ponen en riesgo a los pacientes.

Estudios acelerados y de envejecimiento en tiempo real: Pruebas de Vida Estándar
Las reclamaciones sobre la vida útil de las estanterías son evidentes de que los envases conservan su barrera estéril mientras los dispositivos permanezcan en el mercado. El envejecimiento acelerado proporciona a los fabricantes una visión temprana del rendimiento del embalaje utilizando una temperatura y humedad elevadas para simular años de almacenamiento en apenas semanas. ASTM F1980 (versión actual) delinea el acercamiento, pero la dificultad real es diseñar condiciones que reflejen los ambientes reales sin introducir modos de fallo que nunca ocurrirían en la práctica. Cuando se diseña bien, estos estudios producen datos preliminares que apoyan las reivindicaciones iniciales de protección de la vida útil y ayudan a que los productos avancen mientras que los estudios más largos todavía están en marcha.
El envejecimiento real completa la imagen. Los paquetes de las corridas finales de producción se almacenan en condiciones reales y se supervisan para su rendimiento durante meses o años. Esta evidencia demuestra cómo los envases soportan las realidades cotidianas del almacenamiento y la distribución, siempre que los reguladores de datos confirmados esperen. Los equipos que empiezan estudios en tiempo real antes de tiempo evitan brechas de datos que pueden ralentizar las aprobaciones o renovaciones después.
Las determinaciones más fiables de la vida útil provienen de la combinación de proyecciones aceleradas con confirmación en tiempo real. El análisis estático muestra cuando las propiedades de empaquetado caen por debajo de sus objetivos y le da confianza para los reclamos de vida de la protección. La planificación débil, sin embargo, puede socavar todo el esfuerzo: probar prototipos en lugar de realizar carreras de producción, seleccionar condiciones de almacenamiento que no reflejan la realidad, o subestimar tamaños de muestra erosiona la credibilidad.
Con el enfoque correcto, estudios que envejecen demuestran que los envases protegerán a los pacientes a lo largo de su vida útil, al tiempo que dan a los fabricantes y a los reguladores la confianza de que las afirmaciones se mantendrán con el tiempo. Esta evidencia va más allá de los requisitos reglamentarios para demostrar el desempeño del mundo real que importa para la seguridad de los pacientes.
Evaluación de utilidades y factores humanos: Validar la interacción del usuario
Un sistema de empaquetado puede cumplir con todos los estándares técnicos y todavía fallar si no se puede abrir de forma segura en manos de un clínico. La validación de usabilidad asegura que los sistemas de barrera estériles no sólo contengan en el laboratorio, pero también en el momento del mundo real en el que se abren los dispositivos para el cuidado de los pacientes. Tanto la FDA como la ISO 11607 vinculan la usabilidad directamente a la gestión de riesgos, reconociendo que el embalaje que compromete la técnica aséptica es efectivamente un sistema fallido.
Un enfoque central es pruebas asépticas de presentación - prueba de que la barrera estéril puede ser abierta de forma segura en la práctica. Los sistemas de cierre y las juntas de embalaje deben resistir la insuficiencia prematura y abrirse sin problemas en condiciones clínicas. La integridad de los sellos es esencial. Los paquetes deben pelarse limpiamente sin desgarrar y el diseño debe mantener las partículas o los desechos fuera del campo estéril. Un problema frecuente es el sello con exceso de ingeniería - lo suficientemente fuerte como para cumplir con los objetivos de las pruebas, pero tan difícil de abrir que la técnica aséptica se vuelve poco práctica. Las pruebas de uso simuladas ayudan a superar estos problemas antes de que los productos lleguen al mercado.
En la práctica, las pruebas a menudo implican enfermeras o técnicos quirúrgicos que abren paquetes en condiciones estériles, mientras que los observadores observan cualquier ruptura técnica. El tipo de guante importa: el nitrilo y el látex proporcionan diferentes agarre y sensación. Otros factores del mundo real - como la iluminación O, presión del tiempo durante los procedimientos, o hasta qué punto el paquete en sí mismo coopera - también afecta la capacidad de mantener la esterilidad. Incluso detalles como las pestañas de piel pueden marcar la diferencia: fácil de manejar con las manos desnudas, pero casi imposible con los guantes. La excesiva fuerza de las focas plantea el mismo riesgo, obligando a surgir problemas que socavan todo el propósito de los envases estériles. Un sello que no se puede abrir asépticamente no sólo frustra a los usuarios, sino que compromete directamente la esterilidad en el momento de la atención.
Igualmente importante es entender cómo interactúan las personas con el paquete en la práctica. Estudios de uso observan que los proveedores de atención médica abren dispositivos en condiciones realistas, superando momentos de vacilación junto con la variación técnica o el riesgo potencial de contaminación. Estas observaciones revelan problemas que faltan pruebas materiales - sellos que exigen fuerza excesiva o embalaje que se desgarra imprevisiblemente cuando los usuarios necesitan acceso rápido. Las señales de apertura poco claras crean confusión que comprometen la técnica estéril durante los procedimientos reales.
Finally, validation must account for training requirements. Si el uso seguro depende de instrucciones complejas, aumenta el riesgo de error. La evaluación de las necesidades de formación ayuda a confirmar que el embalaje es intuitivo para sus usuarios previstos. Cualquier instrucción esencial para un manejo seguro debe ser documentada con claridad. Los equipos que toman este paso hacia delante están mejor posicionados para evitar costosos rediseños o reclamaciones inesperadas después del lanzamiento.
Cuando la usabilidad está incluida en la validación desde el principio, los fabricantes ganan más que la evidencia de cumplimiento. Entregan embalajes que funcionan tanto para pacientes como para médicos, al tiempo que aseguran que los reguladores actúen donde más importa.
Cualificación del rendimiento del proceso: Validación de Consistencia de Manufacturing
Incluso el paquete más cuidadosamente diseñado falla si el proceso de fabricación no puede ofrecer el mismo resultado cada vez. La cualificación del rendimiento de los procesos (PPQ) demuestra que los envases no sólo tienen éxito en el laboratorio o durante los ensayos de validación, sino que siguen siendo consistentes a escala de producción completa. Este es el punto en el que tanto los reguladores como los equipos internos de calidad necesitan pruebas de que los sistemas de embalaje pueden soportar la producción del mundo real.
Un fuerte PPQ examina toda la secuencia de producción, incluyendo los procesos de ensamblaje. Analiza cómo se almacenan los materiales, cómo los operadores manejan los componentes y si los controles medioambientales mantienen estables las condiciones. Cualquier debilidad en esas áreas puede erosionar el rendimiento del embalaje, incluso cuando se marcan los ajustes del equipo.
La fiabilidad proviene de la repetibilidad. Por eso PPQ requiere varias ejecuciones de producción para probar que el proceso ofrece resultados consistentes en diferentes lotes. Los equipos típicamente validan tres lotes consecutivos para demostrar que el rendimiento es consistente en diferentes carreras de producción, aunque los requisitos pueden variar en función del riesgo del producto y de la orientación regulatoria. El seguimiento continuo sostiene entonces esa seguridad, capturando la deriva antes de que se convierta en cuestiones de calidad.
Las ganancias van mucho más allá de la conformidad. Un PPQ bien ejecutado ofrece beneficios que llegan muy lejos de las operaciones diarias. Hace que la producción sea más predecible al tiempo que acelera las decisiones de liberación. Lo más importante es que crea confianza con los reguladores en que el sistema está bajo control.
Requisitos de evaluación y gestión de cambios
El cambio es inevitable en la fabricación y cada cambio plantea la cuestión de si su sistema de envases sigue haciendo su trabajo. La evaluación proporciona la evidencia de que lo hace, evitando que pequeños ajustes generen grandes riesgos. Los reguladores ven el cambio incontrolado como una bandera roja, y los fabricantes también deberían hacerlo. Los disparadores comunes incluyen:
- Actualizaciones de equipos, tales como nuevos sistemas de sellado o inspección
- La esterilización cambia, ya sea un cambio en los ajustes de método o parámetro
- Modificaciones de materiales, desde cambiar proveedores hasta seleccionar diferentes sustratos
- El proceso cambia, incluso los menores que alteran las condiciones de funcionamiento
The scope of revalidation depends on risk and documented validation requirements. Any change to a process raises the question of sterility. Los equipos pueden encontrar que un pequeño ajuste afecta a la integridad del sello. En otros casos, es necesario reevaluar la compatibilidad material. A veces la preocupación es si las propiedades de la barrera microbiana seguirán manteniéndose. Cualquiera que sea el activador, las pruebas de seguimiento proporcionan la prueba de que el embalaje sigue protegiendo según lo deseado. También se esperan revisiones regulares, ya que los equipos se desgastan con el paso del tiempo. Los cambios en los proveedores o la deriva gradual del proceso pueden tener el mismo impacto, y cada uno debe ser controlado.
Una buena documentación agrupa todo esto en la confianza de los reguladores de la evidencia. Debería explicar el razonamiento detrás de cada prueba, no sólo capturar las pruebas. Los registros claros proporcionan más que un rastro de papel. Dan confianza a los reguladores, ayudan a los equipos a gestionar el cambio de forma proactiva y, lo que es más importante, muestran que el embalaje sigue protegiendo a los pacientes mucho más allá del primer informe de validación.
Desafíos comunes y soluciones efectivas
La validación puede tropezar por razones familiares. Los tamaños de muestra son a menudo subestimados, las pruebas del peor caso se omiten y la documentación se desliza hasta que ya no refleja lo que se ha hecho. Los equipos también pasan horas comparando documentos de forma manual y adivinando si han detectado cada cambio crítico. Cada uno de estos errores puede sentirse pequeño en el momento, pero juntos crean costosos cambios de trabajo e invitan a hallazgos regulatorios que dañan la credibilidad mucho después de que finalice la auditoría. El cuello de botella de revisión manual es especialmente problemático porque reduce a profesionales de calidad experimentados en comparaciones tediosas en lugar de aplicar su experiencia a la evaluación de riesgos y la toma de decisiones estratégicas.
El enfoque más sólido es construir la validación como una actividad estratégica. Los criterios de aceptación claros establecidos desde el principio ayudan a prevenir desacuerdos en etapas tardías, especialmente cuando los equipos de calidad, ingeniería y regulación se alinean a principios del proceso. Cuando la validación corre a lo largo del desarrollo en lugar de ser tratada como un paso final, las brechas se vuelven más fáciles de detectar mientras que las revisiones reguladoras se mueven más suavemente.
Trabajar eficientemente significa poner el esfuerzo donde más cuenta, no sólo cortar maíz. La muestreo basado en riesgos se centra en recursos en los que el fracaso sería más importante. Los planes de ensayo pueden ser diseñados para satisfacer más de un requisito a la vez, recortando redundancia sin cortar rigor. Utilizado de esta manera, las mejores prácticas ahorran tiempo mientras fortalecen la base de pruebas, asegurando que los envases ganan la confianza del regulador y continúan protegiendo a los pacientes en uso.
Documentación y presentación regular
Para los reguladores, si no está documentado, no sucedió. Los fabricantes se enfrentan a la misma realidad: los registros incompletos pueden ralentizar las aprobaciones mientras desatan los resultados de las inspecciones que debilitan la confianza en el sistema de calidad.
Una documentación sólida cierra esas lagunas. Un registro completo muestra cómo se planificaron y ejecutaron las pruebas mientras conectaban los resultados con los criterios de aceptación. Esta documentación demuestra cumplimiento reglamentario. Los certificados de validación y los archivos técnicos formalizan esta evidencia, confirmando que los sistemas de embalaje cumplen con las expectativas ISO 11607 y FDA. Más que un enlace de informes, la documentación efectiva reúne progenitores, datos y evaluaciones de riesgos en una historia de conformidad controlada y lista para la inspección. Cuando los reguladores preguntan, los fabricantes tienen que mostrar no sólo lo que se ha probado, sino por qué, cómo y con qué resultado.
Una documentación minuciosa no sólo satisface a los reguladores, sino que mantiene los dispositivos que se desplazan al mercado sin interrupciones costosas. Cuando los registros están completos y listos para la inspección, los fabricantes pueden defender su proceso con confianza y los pacientes reciben embalajes que los protege a lo largo del ciclo de vida del dispositivo.
Asegurar el éxito del sistema de empaquetado
La validación del embalaje demuestra que los sistemas de barrera estériles pueden soportar el viaje desde el suelo de la fábrica hasta el punto de cuidado. La validación se basa en una planificación cuidadosa y respaldada por sólidas pruebas y documentación, lo que da confianza a los fabricantes en que su embalaje funcionará en el campo y satisfará a los reguladores. Tratar la validación como una responsabilidad del ciclo de vida, no como un ejercicio único, previene los costosos fracasos y fortalece la confianza entre equipos y reguladores de calidad por igual.
El paisaje está evolucionando. Las plataformas digitales de validación y los vínculos más estrechos con la gestión de riesgos están reformulando las mejores prácticas, mientras que los reguladores avanzan hacia una mayor alineación global. Lo que no cambiará es el objetivo: asegurar que los envases mantengan la esterilidad para que los médicos puedan confiar en todos los dispositivos y los pacientes reciban la protección que merecen. De cara al futuro, una mayor integración de la validación de los envases con la validación de la esterilización dará forma a la práctica del sector, asegurando que las reclamaciones de esterilidad sean apoyadas de extremo a extremo.
La verificación de GlobalVision acelera la validación del empaquete, automatizando la inspección de documentos y diseños, dando confianza a los equipos en el cumplimiento sin ralentizarlos.
