
Quando un dispositivo medico sterile fallisce in chirurgia perché il suo imballaggio non poteva mantenere la sterilità durante la spedizione, la sicurezza del paziente soffre prima. I richiami del prodotto e la reputazione dell'azienda danneggiata seguono da vicino dietro.
Introduzione alla convalida degli imballaggi
La convalida dell'imballaggio del dispositivo medico dimostra che i vostri sistemi di imballaggio proteggono la sterilità del dispositivo dalla produzione fino al punto di cura. La confezione funge da sistema sterile di barriera che mantiene la protezione microbica durante la sterilizzazione e la distribuzione, aiutando i dispositivi a rimanere sterili per anni prima di raggiungere i pazienti.
ISO 11607 fornisce il quadro internazionale per la convalida di questi sistemi. La parte 1 stabilisce requisiti per i materiali e i sistemi sterili di barriera utilizzati per imballare dispositivi medici sterilizzati terminalmente. La parte 2 fornisce i requisiti di convalida per la formatura, la sigillatura e i processi sterili del sistema di barriera. Insieme, garantiscono che il confezionamento sia in grado di mantenere la sterilità e dimostrato di funzionare in condizioni di produzione del mondo reale. Il regolamento sulla qualità del sistema FDA (21 CFR 820) impone requisiti paralleli attraverso i controlli di progettazione e i mandati di convalida del processo, rendere necessaria la convalida della conformità piuttosto che una misura facoltativa di qualità.
La convalida è tipicamente guidata dalla garanzia della qualità, ma il successo reale dipende dal coordinamento tra i vari reparti. Molti ritardi derivano dalla confusione su ruoli o lacune nelle aspettative. Quando i gruppi di ingegneria, regolamentazione e produzione contribuiscono alla loro esperienza sincronizzata, la convalida diventa molto più efficiente e affidabile.
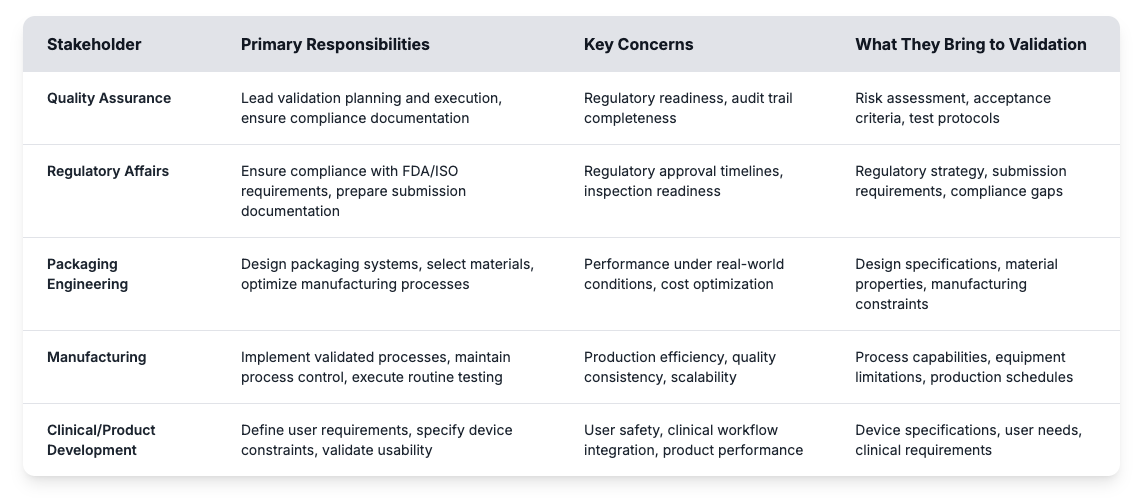
Ottenere questi ruoli giusti aiuta i produttori di dispositivi medici a costruire programmi di convalida efficaci ed evitare insidie comuni. La valutazione dei rischi individua potenziali punti di insuccesso mentre i criteri di accettazione stabiliscono obiettivi misurabili per le prestazioni degli imballaggi. I protocolli di prova definiscono il modo in cui i test di convalida dimostreranno che i sistemi sterili a barriera soddisfano i requisiti regolamentari per tutta la durata di validità prevista. Costruire un programma di convalida di successo inizia con la comprensione di questi requisiti fondamentali e la definizione di processi chiari che collegano input degli stakeholder ai risultati misurabili.
Requisiti Fondazionali Per La Convalida Dell'Imballaggio
Ogni programma di convalida di successo inizia con tre elementi di base della convalida dell'imballaggio che i regolatori si aspettano di vedere:
- Piano di convalida completo: Il tuo piano di convalida collega le decisioni di progettazione alla documentazione di conformità. Definisce chi fa cosa, quando i test accade, e come documenterai i risultati. I regolatori esaminano questi piani durante le ispezioni per vedere se il vostro approccio ha senso o se state semplicemente controllando scatole. I buoni piani comprendono protocolli chiari con compiti di responsabilità e scadenze realistiche che tengono conto dei potenziali ritardi.
- Valutazione strutturata del rischio: La valutazione del rischio ti aiuta a focalizzare le risorse di convalida dove sono più importanti. Identificherai gli scenari in cui l'imballaggio potrebbe compromettere la sterilità del dispositivo o la sicurezza - cose come i guasti della tenuta durante la spedizione, problemi di materiale con il metodo di sterilizzazione, o imballaggio che è troppo difficile per gli utenti di aprirsi in modo sicuro. Questa valutazione ti dice quali test sono critici e quali sono belli da avere.
- Criteri di accettazione scientificamente giustificati: I criteri di accettazione definiscono come "abbastanza buoni" le prestazioni dell'imballaggio. Essi devono essere specifici e misurabili pur rimanendo legati alla sicurezza dei pazienti piuttosto che alla convenienza della fabbricazione. Invece di criteri vaghi come "aspetto accettabile", fisserai obiettivi per la forza della sigilla, tassi di perdite, e manutenzione di sterilità che hanno senso per il vostro dispositivo specifico e condizioni di utilizzo.
Fondazioni deboli creano problemi di convalida che si moltiplicano come il progresso dei programmi. Le squadre spesso scoprono delle lacune troppo tardi, quando risolverle significa ricominciare con i test che avrebbero dovuto essere inclusi fin dall'inizio. L'effetto domino è prevedibile: la dispersione dei test porta a prove incomplete, il che crea questioni normative che ritardano l'approvazione e aumentano i costi.
I quattro pilastri della convalida degli imballaggi
ISO 11607 rompe la convalida del packaging in quattro pilastri interconnessi che lavorano insieme per dimostrare che il vostro imballaggio non fallirà quando conta di più. Piuttosto che trattarli come caselle di controllo separate, team di successo capire come ogni pilastro si alimenta nel prossimo per costruire prove complete che l'imballaggio protegge la sterilità del dispositivo durante il ciclo di vita del dispositivo.

Non è possibile dimostrare prestazioni di imballaggio coerenti (PQ) senza conoscere il funzionamento dell'apparecchiatura in modo affidabile (OQ). Non puoi fidarti delle prestazioni dell'apparecchiatura senza una corretta installazione (IQ). La qualificazione dei materiali (MQ) si affianca alla convalida delle apparecchiature per garantire che tutto funzioni insieme come un sistema di imballaggio completo. Una volta fatto correttamente, questo approccio sistematico fornisce prove di convalida passo dopo passo con ogni pilastro rafforzando gli altri. Quando salti i passaggi, crei dei problemi costosi:
- Saltare IQ significa che non sai se i problemi di equipaggiamento stanno disordinando i tuoi risultati di test successivi.
- OQ debole ti lascia con parametri di processo che sembrano buoni nei test, ma cadono a parte sotto stress di produzione.
- La qualificazione del materiale da taglio è come i team finiscono con l'imballaggio che fallisce durante la spedizione o perde la sterilità nel tempo.
Pensate alla convalida come costruire una casa - non iniziereste con il tetto prima di posare una solida fondazione. I quattro pilastri funzionano allo stesso modo, con ogni fase a sostegno di ciò che verrà dopo. Salta il terreno e passerai molto più tempo e denaro per risolvere i problemi che avrebbero potuto essere evitati con un approccio sistematico fin dall'inizio.
Qualificazione dell'installazione: Verifica e taratura dell'apparecchiatura
Senza una solida documentazione di base, i problemi di apparecchiature e i guasti di processo diventano impossibili da separare nelle fasi successive di convalida. La qualificazione di installazione (IQ) impedisce questo risultato stabilendo esattamente cosa stai iniziando prima che inizi qualsiasi test di prestazioni.
Il core deliverable da IQ è la documentazione dei parametri di base che cattura il punto di partenza dell'apparecchiatura. Registrerai:
- Intervalli di funzionamento per parametri critici come temperatura, pressione, velocità e tempo di permanenza
- Impostazioni predefinite e configurazioni di sistema di controllo
- Condizioni ambientali nella zona di imballaggio
- Caratteristiche di prestazione dell’apparecchiatura in condizioni a vuoto
- Requisiti di manutenzione preventiva e programmi che mantengono le apparecchiature in modo coerente
La verifica e la taratura delle apparecchiature creano le basi per queste linee di base. Confermerai che tutti i componenti corrispondono alle specifiche di acquisto, le utenze soddisfano i requisiti e i sistemi di sicurezza funzionano correttamente. Per le attrezzature di sigillatura, documenterai le capacità di temperatura e pressione insieme alle specifiche degli elementi di riscaldamento. I sistemi di controllo vengono verificati per confermare che possono mantenere i parametri richiesti.
La taratura estende la fondazione del QI. Strumenti che controllano o controllano il processo di confezionamento - regolatori di temperatura, manometri sistemi di tempistica e strumenti di prova - devono disporre di certificati riconducibili alle norme nazionali. Tali registrazioni registrano le prestazioni delle apparecchiature e dimostrano il controllo di processo durante la convalida e la produzione di routine.
Tale record diventa anche il punto di riferimento per le fasi successive. Se la resistenza alla tenuta si sposta durante la qualificazione delle prestazioni, i risultati possono essere ricondotti alla variazione dell'apparecchiatura documentata in QI. Senza questo livello di documentazione, distinguere tra problemi di apparecchiature e guasti di processo diventa difficile, portando a costose indagini che un accurato lavoro iniziale avrebbe evitato.
Qualificazione Operativa: Testing Across Operating Ranges
Il vostro equipaggiamento potrebbe funzionare perfettamente in condizioni ideali, ma dare risultati incoerenti quando la produzione diventa disordinata. La qualificazione operativa (OQ) rivela queste zone di difficoltà prima che causino problemi a valle. Testando i parametri ai loro limiti, i team possono vedere dove il processo si mantiene stabile e dove le prestazioni cominciano a rompere.
Il vostro obiettivo è stabilire i parametri di processo che i team di produzione utilizzeranno quotidianamente: i limiti operativi con i normali set point più intervalli di variazione accettabili. Troverete gamme in cui l'attrezzatura produce risultati affidabili in modo coerente, con impostazioni ottimali che danno le migliori prestazioni e margini di sicurezza che evitano problemi. La funzionalità delle apparecchiature di prova significa variare sistematicamente un parametro mantenendo gli altri costanti, quindi monitorare gli output di qualità dei pacchetti. La prova ai bordi aiuta a identificare dove inizia la rottura delle prestazioni. Questo diventa il vostro margine di sicurezza per la produzione di routine.
I record OQ devono andare oltre l'elenco di ciò che l'apparecchiatura può fare. Essi dovrebbero anche catturare le condizioni che mantengono i risultati coerenti. Ad esempio, elevata umidità può interferire con la sigillatura, cambiamenti di temperatura può causare deriva nei parametri critici e macchinari vicini potrebbe introdurre vibrazioni che riducono la precisione.
Questo test cattura combinazioni di parametri che creerebbero problemi di produzione prima che si presentino effettivamente. Il tuo equipaggiamento potrebbe funzionare a determinate impostazioni ma dare risultati incoerenti che causano mal di testa a valle. OQ identifica queste zone di difficoltà in modo che le squadre di produzione possano guidare chiaramente, con le gamme che stabilisci qui diventando la base per l'addestramento dell'operatore e gli allarmi di processo che continuano a produrre in modo fluido.
Prestazioni Qualificazione: Prestazioni Di Imballaggio Coerenti
La qualificazione delle prestazioni (PQ) dimostra che il processo di imballaggio completo produce costantemente pacchetti che soddisfano tutti i criteri di accettazione in condizioni di produzione reali. Qui è dove la convalida si sposta dalla capacità di dimostrare l'affidabilità.
La realtà è che il vostro processo di imballaggio deve funzionare quando tutto non è perfetto. Le impostazioni delle apparecchiature si spostano leggermente, gli operatori cambiano tra i turni e i lotti di materiale variano di qualità. Le condizioni ambientali fluttuano per tutta la giornata. I test della MP mostrano che il processo produce ancora pacchetti accettabili nonostante queste normali variazioni. Il peggiore scenario test lo porta avanti spingendo il processo convalidato a limiti realistici. Le prove di stress dovrebbero riflettere le realtà della produzione, non solo le condizioni ideali. Ciò può significare funzionare alla velocità massima della linea mentre gli operatori spingono per rispettare le scadenze, lavorare con controlli ambientali ridotti se i sistemi HVAC non funzionano bene, o testare la qualità alla fine di uno spostamento quando la fatica può influenzare i risultati.
Prova di convalida in esecuzione in condizioni di produzione effettive significa utilizzare gli stessi materiali, impostazioni delle apparecchiature e condizioni ambientali che i team di produzione devono affrontare quotidianamente. Catturare le variazioni naturali di processo nel tempo, operatori, lotti di materiali e attrezzature afferma conta più che collaudare in condizioni artificialmente controllate che non rappresentano la realtà. La pianificazione statistica stabilisce la quantità di prove necessarie per dimostrare prestazioni coerenti. Un piano di campionamento ben progettato copre più corse e operatori, assicurando che i risultati riflettano la capacità di processo reale invece che il caso. Questa prova quindi guida il monitoraggio del processo e sostiene le prestazioni convalidate durante la produzione quotidiana.
Materiali e Design Qualificazione: Garantire la Compatibilità e la Protezione
La qualificazione del materiale (MQ) mostra che i materiali di imballaggio del dispositivo possono funzionare come previsto per tutto il ciclo di vita del dispositivo. Per i dispositivi medici sterilizzati terminali, questo significa selezionare imballaggi primari che resiste al metodo di sterilizzazione scelto e continua a proteggere la sterilità durante la distribuzione e lo stoccaggio. La convalida del sistema di imballaggio conferma quindi che i materiali, la progettazione e la tenuta funzionano insieme come una barriera sterile per tutta la vita prevista del dispositivo.
Nel selezionare i materiali, le squadre devono valutare tre fattori:
- Il metodo di sterilizzazione che verrà utilizzato
- L'ambiente di archiviazione al quale il pacchetto deve resistere
- I requisiti dell'utente per l'apertura sicura e l'accesso al dispositivo
Ogni metodo di sterilizzazione presenta sfide uniche. La sterilizzazione dell'ossido di etilene richiede tipicamente materiali che resistono alla degradazione chimica consentendo la penetrazione e l'evacuazione del gas per una sterilizzazione e un'aerazione efficaci. La sterilizzazione gamma espone l'imballaggio a radiazioni che possono indebolire la forza e la flessibilità. La sterilizzazione a vapore combina l'alta temperatura con l'umidità che può compromettere le proprietà della barriera. Se questi rischi non vengono affrontati presto, la barriera sterile può fallire nell'uso reale, mettendo a rischio sia la conformità e la sicurezza dei pazienti.
Il test di compatibilità dei materiali valuta come il vostro imballaggio interagisce con i componenti del dispositivo per tutta la durata di conservazione prevista, ma ha anche bisogno di affrontare la pratica usabilità. I materiali devono inoltre mantenere le loro proprietà di barriera microbica per garantire che le barriere sterili continuino a bloccare i contaminanti per tutta la durata di conservazione prevista. Allo stesso tempo, la forza della buccia deve essere abbastanza forte per mantenere l'integrità della foca durante la distribuzione, ma abbastanza gentile per gli operatori sanitari di aprirsi in modo asettico senza lacerare. Si eseguono studi di invecchiamento accelerato che simulano anni di stoccaggio in settimane, combinato con l'invecchiamento in tempo reale in condizioni di conservazione effettive per confermare le tue previsioni.
Protocolli Di Prova Dei Materiali:
| Tipo Di Prova | Valutazioni Principali | Scopo |
|---|---|---|
| Fisico | Resistenza alla guarnizione, prova di scoppio per resistenza alla pressione, prova di colorazione per rilevamento delle perdite | Integrità meccanica |
| Chimico | Estrattili, liscivie, sterilizzanti residui | Sicurezza dei materiali |
| Biologico | Manutenzione della sterilità, biocompatibilità | Sicurezza dei pazienti |
I protocolli di prova iniziali aiutano a stabilire le prestazioni dei materiali di base prima dell'inizio degli studi di convalida completi. Concentrati sulla progettazione di protocolli di prova che stress materiali il modo in cui saranno effettivamente stressati in uso reale piuttosto che soddisfare specifiche arbitrarie di test che non si riferiscono a reali requisiti di prestazioni. Le caratteristiche di apertura dovrebbero consentire un accesso pulito e controllato che non comprometta la sterilità o la dispersione di particelle nel campo sterile, il che significa che i test devono simulare le condizioni cliniche reali di uso piuttosto che gli ambienti di laboratorio ideali.
Test di distribuzione e transito: Simulating Real-World Shipping
L’imballaggio deve resistere al viaggio completo dal pavimento di fabbricazione al letto del paziente. I test di distribuzione ricreano le sollecitazioni del trasporto e della movimentazione in modo che i team possano confermare che le barriere sterili rimangono intatte e funzionali in tutta la catena di fornitura.
I protocolli ASTM D4169 e ISTA delineano come simulare gli ambienti di distribuzione, ma è necessario adattarli alle vostre reali condizioni di spedizione. Invece di controlli isolati, i test dovrebbero riflettere l'intero percorso: un pacchetto può essere eliminato, attillato da vibrazioni di trasporto o schiacciato sotto i carichi di magazzino. Lungo il percorso, spostamenti di orientamento, manipolazione ripetuta e condizioni climatiche mutevoli tutti testare la forza della barriera sterile. L'esecuzione di queste sollecitazioni in sequenza dà un'immagine più fedele di come l'imballaggio funziona nella distribuzione del mondo reale.
I pacchetti passano attraverso diversi controlli dopo il test di distribuzione. Il controllo visivo cerca danni alla superficie, mentre il test di resistenza conferma che le chiusure rimangono intatte. Metodi più sensibili, come i test di bolla, scoprono microperdite in barriere sterili che non possono essere visti dall'occhio. La fase finale è il test di integrità, dimostrare la barriera microbica ancora esegue anche dopo il trasporto simulato - e questi risultati sono importanti solo se il test riflette le condizioni reali di spedizione. I pacchetti che superano i protocolli generici ancora non riescono in campo portano a richiami mentre danneggiano le reputazioni aziendali. Soprattutto, mettono a rischio i pazienti.

Studi di invecchiamento accelerati e in tempo reale: Prove di vita Claims
Le indicazioni sulla durata di validità sono o cadono nella prova che l’imballaggio preserva la sua barriera sterile finché i dispositivi rimangono sul mercato. L'invecchiamento accelerato offre ai produttori una visione precoce delle prestazioni dell'imballaggio utilizzando temperature e umidità elevate per simulare anni di stoccaggio in poche settimane. ASTM F1980 (versione corrente) delinea l'approccio, ma la vera difficoltà è quella di progettare condizioni che riflettano gli ambienti reali senza introdurre modalità di guasto che non si verificherebbero mai nella pratica. Se progettato bene, questi studi producono dati preliminari che supportano le indicazioni iniziali di conservabilità e aiutano i prodotti a progredire mentre sono ancora in corso studi a più lungo termine.
L'invecchiamento in tempo reale completa l'immagine. I colli provenienti da cicli di produzione finale sono immagazzinati in condizioni reali e monitorati per le prestazioni nel corso di mesi o anni. Questa prova dimostra come l'imballaggio resiste alle realtà quotidiane di conservazione e distribuzione, fornendo le aspettative dei regolatori dei dati di conferma. I team che iniziano gli studi in tempo reale evitano precocemente lacune di dati che possono rallentare l'approvazione o il rinnovo in seguito.
Le determinazioni della durata di conservazione più affidabili derivano dalla combinazione di proiezioni accelerate con conferma in tempo reale. L'analisi statistica mostra quando le proprietà di imballaggio cadono sotto i tuoi obiettivi e ti dà intervalli di confidenza per le affermazioni di durata di conservazione. Una pianificazione debole, tuttavia, può minare l'intero sforzo - testare prototipi invece di processi di produzione, selezionare le condizioni di conservazione che non riflettono la realtà, o sottovalutare le dimensioni del campione erodere la credibilità.
Con l'approccio giusto, studi di invecchiamento dimostrano che il confezionamento proteggerà i pazienti durante la durata di vita prevista, dando ai produttori e ai regolatori la fiducia che le affermazioni resteranno nel tempo. Tali prove vanno al di là dei requisiti normativi per dimostrare le prestazioni reali che sono importanti per la sicurezza dei pazienti.
Valutazione dell'usabilità e fattori umani: convalida dell'interazione dell'utente
Un sistema di confezionamento può soddisfare tutti gli standard tecnici e non riuscire se non può essere aperto in modo sicuro nelle mani di un clinico. La validazione dell'usabilità garantisce che i sistemi sterili di barriera non si blocchino solo in laboratorio, ma anche nel momento reale in cui i dispositivi sono aperti per la cura del paziente. Sia FDA che ISO 11607 legano l'usabilità direttamente alla gestione del rischio, riconoscendo che l'imballaggio che compromette la tecnica asettica è effettivamente un sistema fallito.
Un focus centrale è asettico test di presentazione - prova che la barriera sterile può essere aperta in modo sicuro nella pratica. I sistemi di chiusura e le guarnizioni devono resistere a guasti prematuri ma aprirsi senza intoppi in condizioni cliniche. L'integrità del sigillo è essenziale. I pacchetti devono sbucciare in modo pulito senza strappare, e il design deve tenere le particelle o i detriti fuori dal campo sterile. Un problema frequente è il sigillo sovraingegnerizzato - abbastanza forte per raggiungere gli obiettivi di prova, ma così difficile da aprire che la tecnica asettica diventa impraticabile. Il test di utilizzo simulato aiuta la superficie di questi problemi prima che i prodotti raggiungano il mercato.
In pratica, i test spesso coinvolgono infermieri o tecnici chirurgici che aprono pacchetti in condizioni sterili, mentre gli osservatori notano eventuali interruzioni tecniche. Il tipo di guanto contiene: nitrile e lattice forniscono impugnatura differente e sentire. Altri fattori del mondo reale - come l'illuminazione OP, la pressione del tempo durante le procedure, o quanto bene la confezione stessa coopera - influisce anche sulla capacità di mantenere la sterilità. Anche i dettagli come le schede di buccia possono fare la differenza: facile da gestire con le mani nude, ma quasi impossibile con i guanti. La resistenza eccessiva della tenuta presenta lo stesso rischio, costringendo i lavori che compromettono l'intero scopo dell'imballaggio sterile. Un sigillo che non può essere aperto asetticamente non solo frustrare gli utenti, ma compromette direttamente la sterilità nel punto di cura.
Altrettanto importante è capire come le persone interagiscono con il pacchetto nella pratica. Gli studi di utilizzabilità osservano i fornitori di assistenza sanitaria che aprono dispositivi in condizioni realistiche, superando momenti di esitazione insieme a variazioni tecniche o rischio potenziale di contaminazione. Queste osservazioni rivelano problemi che i test del materiale mancano - sigilli che richiedono una forza eccessiva o un imballaggio che lacera imprevedibilmente quando gli utenti hanno bisogno di un accesso rapido. I segnali di apertura poco chiari creano confusione che compromette la tecnica sterile durante le procedure reali.
Infine, la convalida deve tenere conto dei requisiti di addestramento . Se l'uso sicuro dipende da istruzioni complesse, il rischio di errore aumenta. La valutazione precoce delle esigenze di formazione aiuta a confermare che l'imballaggio è intuitivo per i suoi utilizzatori previsti. Tutte le istruzioni essenziali per una manipolazione sicura devono essere chiaramente documentate. I team che prendono questo passo avanti sono posizionati meglio per evitare costose riprogettazioni o reclami inaspettati dopo il lancio.
Quando l'usabilità è integrata nella convalida fin dall'inizio, i produttori ottengono più che prove di conformità. Forniscono imballaggi che funzionano sia per i pazienti che per i clinici, garantendo al contempo i regolatori che eseguono dove conta di più.
Qualificazione Prestazioni Del Processo: Convalida Coerenza Della Produzione
Anche il pacchetto più attentamente progettato fallisce se il processo di produzione non può fornire lo stesso risultato ogni volta. Process Performance Qualification (PPQ) dimostra che gli imballaggi non solo riescono nel laboratorio o durante le prove di convalida, ma rimangono coerenti a piena scala di produzione. Questo è il punto in cui sia i regolatori che i team interni di qualità devono dimostrare che i sistemi di imballaggio possono resistere alla produzione del mondo reale.
Un forte PPQ esamina l'intera sequenza di produzione, compresi i processi di assemblaggio. Si esamina come i materiali vengono immagazzinati, come gli operatori gestiscono i componenti e se i controlli ambientali mantengono le condizioni stabili. Qualsiasi debolezza in queste aree può erodere le prestazioni di imballaggio, anche quando le impostazioni dell'apparecchiatura sono composte.
L'affidabilità deriva dalla ripetibilità. Ecco perché PPQ richiede più fasi di produzione per dimostrare che il processo fornisce risultati coerenti in diversi lotti. I team in genere convalidano tre lotti consecutivi per dimostrare che le prestazioni sono coerenti tra diverse fasi di produzione, anche se i requisiti possono variare in base al rischio del prodotto e agli orientamenti normativi. Il monitoraggio continuo mantiene poi tale garanzia, catturando la deriva prima che si trasforma in problemi di qualità.
Il pagamento va ben al di là del rispetto. Un PPQ ben eseguito offre benefici che raggiungono molto nelle operazioni quotidiane. Rende la produzione più prevedibile, accelerando le decisioni di rilascio. Soprattutto, crea fiducia con i regolatori nel fatto che il sistema è sotto controllo.
Requisiti di rivalidazione e gestione del cambiamento
Il cambiamento è inevitabile nella produzione, e ogni cambiamento solleva la questione se il vostro sistema di imballaggio fa ancora il suo lavoro. La rivalidazione fornisce la prova che lo fa, impedendo che piccoli aggiustamenti creino grandi rischi. I regolatori considerano il cambiamento incontrollato come una bandiera rossa, e anche i produttori dovrebbero farlo. I trigger comuni includono:
- Aggiornamenti delle apparecchiature, come nuovi sistemi di sigillatura o di ispezione
- La sterilizzazione cambia, sia che si tratti di una modifica del metodo o di un parametro
- Modifiche del materiale, dal passaggio di fornitori alla selezione di substrati diversi
- Processo cambia, anche quelli minori che alterano le condizioni operative
La portata del rinnovo dipende dai requisiti di rischio e di convalida documentata.Qualsiasi modifica ad un processo solleva la questione della sterilità. Le squadre possono scoprire che una piccola regolazione influisce sull'integrità della guarnizione. In altri casi, la compatibilità materiale deve essere riesaminata. A volte la preoccupazione è se le proprietà di barriera microbica resteranno ancora. Qualunque sia l'innesco, i test di follow-up forniscono la prova che l'imballaggio continua a proteggere come previsto. Sono previste anche recensioni regolari, dal momento che le attrezzature indossano nel tempo. I trasferimenti di fornitori o la deriva graduale del processo possono avere lo stesso impatto e ciascuno di essi deve essere monitorato.
Una buona documentazione tira tutto questo insieme in autorità di regolamentazione delle prove può fidarsi. Dovrebbe spiegare il ragionamento alla base di ogni test, non solo catturare che i test si sono verificati. I record chiari forniscono più di un percorso cartaceo. Danno fiducia ai regolatori, aiutano i team a gestire il cambiamento in modo proattivo e, soprattutto, mostrano che il confezionamento continua a proteggere i pazienti ben oltre il primo rapporto di convalida.
Sfide comuni e soluzioni efficaci
La convalida può inciampare per motivi familiari. Le dimensioni del campione sono spesso sottovalutate, i test peggiori vengono saltati e la documentazione scivola fino a quando non riflette più ciò che è stato fatto. I team trascorrono anche ore a confronto manuale dei documenti e a seconda ipotesi se hanno colto ogni cambiamento critico. Ognuno di questi errori può sentirsi piccolo nel momento, ma insieme creano costosi lavori di revisione e invitano a riscontri normativi che danneggiano la credibilità molto tempo dopo la conclusione dell'audit. La strozzatura della revisione manuale è particolarmente problematica, in quanto colpisce professionisti di qualità esperti in confronti noiosi invece di applicare la loro competenza alla valutazione del rischio e al processo decisionale strategico.
L'approccio più forte è quello di costruire la convalida come attività strategica. I chiari criteri di accettazione stabiliti fin dall'inizio aiutano a prevenire disaccordi in fase avanzata, soprattutto quando i team di qualità, ingegneria e regolamentazione si allineano nelle fasi iniziali del processo. Quando la convalida avviene durante tutto lo sviluppo piuttosto che essere trattata come una fase finale, le lacune diventano più facili da individuare mentre le revisioni regolamentari procedono più agevolmente.
Lavorare in modo efficiente significa impegnare dove conta di più, non solo tagliando gli angoli. Il campionamento basato sul rischio si concentra sulle risorse in cui il fallimento sarebbe più importante. I piani di prova possono essere progettati per servire più di un requisito alla volta, ridondanza di taglio senza tagliare rigoroso. Usato in questo modo, le migliori pratiche risparmiano tempo rafforzando la base delle prove, garantendo che l'imballaggio guadagni la fiducia del regolatore e continua a proteggere i pazienti in uso.
Documentazione e presentazione normativa
Per i regolatori, se non è documentato, non è accaduto. I produttori affrontano la stessa realtà: i record incompleti possono lentare le approvazioni mentre innescano i risultati delle ispezioni che indeboliscono la fiducia nel sistema di qualità.
Una forte documentazione colma tali lacune. Un record completo mostra come i test sono stati pianificati ed eseguiti durante la connessione dei risultati ai criteri di accettazione. Questa documentazione dimostra la conformità alla normativa. I certificati di convalida e i file tecnici formalizzano quindi questa prova, confermando che i sistemi di imballaggio soddisfano le aspettative ISO 11607 e FDA. Più che un raccoglitore di relazioni, una documentazione efficace intreccia protocolli, dati e valutazioni dei rischi in una storia di conformità controllata e pronta per l'ispezione. Quando i regolatori chiedono, i produttori devono dimostrare non solo ciò che è stato testato, ma perché, come e con quale risultato.
Una documentazione accurata non solo soddisfa i regolatori; mantiene i dispositivi spostati sul mercato senza costose interruzioni. Quando i record sono completi e pronti per l'ispezione, i produttori possono difendere il loro processo con fiducia e i pazienti ricevono imballaggi che li proteggono durante il ciclo di vita del dispositivo.
Garantire il successo del sistema di imballaggio
La convalida degli imballaggi dimostra che i sistemi sterili di barriera possono resistere al viaggio dal pavimento della fabbrica al punto di cura. Messa a punto in una pianificazione attenta e supportata da prove e documentazione solide, la convalida dà fiducia ai produttori il loro imballaggio eseguirà sul campo e soddisfare i regolatori. Trattare la convalida come responsabilità del ciclo di vita, non come un esercizio una tantum, prevenire disfunzioni costose e rafforzare la fiducia tra team di qualità e autorità di regolamentazione.
Il paesaggio è in evoluzione. Piattaforme di convalida digitale e collegamenti più stretti alla gestione del rischio stanno rimodellando le migliori pratiche, mentre le autorità di regolamentazione si muovono verso un maggiore allineamento globale. Quello che non cambierà è l'obiettivo: garantire che il confezionamento mantenga la sterilità in modo che i medici possano fare affidamento su ogni dispositivo e i pazienti ricevano la protezione che meritano. Guardando al futuro, una più stretta integrazione della convalida degli imballaggi con la convalida della sterilizzazione formerà la pratica del settore, garantendo che le rivendicazioni di sterilità siano supportate end-to-end.
GlobalVision’s Verify accelera la convalida del packaging automatizzando l'ispezione di documenti e opere d'arte, dando ai team la sicurezza della conformità senza rallentarli.
